Introduction
Left ventricular hypertrophy (LVH) is a condition in which there is an increase in left ventricular mass, with or without an increase in chamber size (see Image. Specimen Showing Myocardial Infarction).[1] The left ventricle is the chamber responsible for pumping oxygenated blood to the rest of the body, and hypertrophy can occur as a response to increased workload or other conditions. This condition is often asymptomatic in early stages, but advanced disease is associated with advanced cardiac pathologies. If untreated, LVH can lead to heart failure, arrhythmias, ischemia, or stroke. Management is multifactorial and includes treating underlying conditions, lifestyle modification, drug therapy, device therapy, and surgical or catheter-based interventions, depending on disease pathology.
LVH can manifest in 2 distinct forms, concentric and eccentric, depending on the underlying cause and the type of cardiac workload (pressure or volume overload).[1] In concentric hypertrophy, wall thickness increases symmetrically, and the sarcomeres are added in parallel. Concentric LVH occurs in conditions of pressure overload, such as aortic stenosis. In eccentric LVH, wall thickness increases along with chamber dilation. The sarcomeres are added in series. Eccentric LVH occurs in conditions of volume overload, such as aortic regurgitation. LVH can also be classified based on the pattern of hypertrophy, ie, symmetrical or asymmetrical.
LVH is secondary to conditions increasing cardiac workload. The closely relevant term 'hypertrophic cardiomyopathy' (HCM) refers to a primary genetic condition characterized by hypertrophy of the heart muscle, particularly in the left ventricle. Mutations in sarcomeric protein genes cause this condition. Several diagnostic features help distinguish the 2 entities.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Causes of concentric hypertrophy include:
- Essential hypertension [2]
- Valvular aortic stenosis
- Coarctation of the aorta
- Renal artery stenosis
- Subaortic stenosis (left ventricular outflow tract obstruction by muscle or membrane)
- Cardiac amyloidosis
- Anderson-Fabry disease
Causes of eccentric hypertrophy include:
- Mitral regurgitation
- Aortic regurgitation
- Chronic kidney disease
- Ventricular septal defect
- Dilated cardiomyopathy
Physiological causes include:
- Athlete heart
- An adaptive hypertrophy to meet increased cardiovascular demands
Genetic conditions:
- HCM
- Infiltrative disorders like amyloidosis, Pompe disease (glycogen storage disorder), or Fabry disease (lysosomal storage disorder)
Other causes:
- Obesity
- Diabetes mellitus
- Coronary artery disease
- Coronary artery disease has been shown to contribute to the pathogenesis of LVH, as the normal myocardium compensates for ischemic or infarcted tissue.
- Anemia
- Hyperthyroidism
Epidemiology
LVH, which is associated with a high incidence and risk of death from cardiovascular disease, is thought to affect more than 30% of patients with hypertension globally.[3] This condition is more often prevalent in Black populations, older adults, individuals with obesity, and patients with hypertension.[4] A review of echocardiographic data of 37,700 individuals revealed 19% to 48% prevalence of LVH in untreated hypertensives and 58% to 77% in high-risk hypertensive individuals.
The presence of obesity also causes a 2-fold increased risk of developing LVH. The prevalence of LVH ranges from 36% (conservative criteria) to 41% (less conservative criteria) in the population, depending on the criteria used for defining it. LVH prevalence is not reported to differ between men and women, with estimates ranging from 36.0% to 37.9% (conservative criteria) and 43.5% to 46.2% (less conservative criteria). The prevalence of eccentric LVH is relatively higher compared to concentric hypertrophy.[4]
Pathophysiology
Stimulus for Hypertrophy
LVH and remodeling, early on, are important compensatory processes that develop over time in response to wall stress, significant hemodynamic pressure, or volumetric burden. The increased mass of muscle fibers or wall thickness initially serves as a compensatory mechanism to maintain contractile forces and counter increased ventricular wall stress. The benefits of increased wall thickness to compensate for elevated wall stress are offset by a significant increase in the degree of stiffness of the hypertrophied walls associated with a significant increase in diastolic ventricular pressures, which are subsequently transmitted back into the left atrium as well as the pulmonary vasculature.
Pathologic LVH, once developed, puts the patient at significant risk for the development of heart failure, dysrhythmias, and sudden death. The most common etiologic cause is the heart contracting against an elevated afterload, as seen in hypertension and also seen in valvular aortic stenosis. Another cause is increased filling of the left ventricle, inducing diastolic overload, which is the underlying mechanism for eccentric LVH in patients with regurgitant valvular lesions, such as aortic regurgitation or mitral regurgitation, and also seen in dilated cardiomyopathy. Coronary artery disease has been shown to contribute to the pathogenesis of LVH, as the normal myocardium compensates for ischemic or infarcted tissue. This type of sustained increase in wall stress, along with cytokine and neuroactivation, stimulates the development of myocardial hypertrophy, or an increase in muscle thickness, with deposition of the extracellular matrix.
Concentric LVH is an abnormal increase in left ventricular myocardial mass resulting from pressure overload induced by arteriolar vasoconstriction, as occurs in chronic hypertension or aortic stenosis. Eccentric LVH is induced by an increased filling pressure of the left ventricle, otherwise known as diastolic overload, which represents the underlying mechanism for volumetric or diastolic overload in patients with regurgitant valve lesions, such as aortic or mitral regurgitation, as well as in the case of dilated cardiomyopathy. In patients with coronary artery disease, these mechanisms can play a role in an attempt to compensate for ischemic or infarcted myocardial tissue.[5]
Myocardial Fibrosis
One key pathophysiologic component in LVH is the concomitant development of myocardial fibrosis. Initially, fibrosis is clinically manifested by diastolic dysfunction, but systolic dysfunction will also develop with progressive disease.[6] Myocardial fibrosis appears to be pathophysiologically linked to the renin-angiotensin-aldosterone system.
Evidence has been established that angiotensin II produces a profibrotic effect in the myocardial tissue of hypertensive individuals. This explains why angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are among the most potent agents in the treatment of hypertension, especially from the standpoint of morbidity and mortality. LVH is a consistent predictor of cardiovascular morbidity as well as mortality in hypertensive individuals.[7] Certain antihypertensive therapies that induce regression of LVH decrease rates of major adverse cardiovascular events and enhance survival, regardless of the degree of blood pressure reduction. The clinical importance is 2-fold: recognizing that LVH can be a modifiable risk factor and that management choices are significantly more complex than simply controlling blood pressure.[8]
Molecular Signaling Pathways
Various signaling modulators in the cardiovascular system have been shown to regulate cardiac mass, including effects on gene expression, apoptosis, cytokines, and growth factor stimulation. Recent studies suggest that pathological hypertrophy can be reversed or prevented altogether. Other molecular mechanisms that may cause cardiac hypertrophy include the aberrant re-expression of a fetal gene program.
A variety of molecular pathways that potentially coordinate the control of the hypertrophic program include the following: natriuretic peptides, the adrenergic nervous system, adhesion and cytoskeletal proteins, interleukin-6 cytokines, MEK-ERK1/2 signaling, histone acetylation, calcium-mediated modulation, and microRNAs. At the cellular level, cardiomyocyte hypertrophy is characterized by increased cell size, upregulated protein synthesis, and intensified sarcomere organization. Mechanical stress induces a hypertrophic response downstream of mechanosensitive molecular structures.
The sarcomeric Z-disc and its proteins seem to drive mechanical stress-induced signal transduction or mechanotransduction. Examples of mechanosensitive molecules include a family of Z-disc-specific proteins, including calsarcins and myozenins. Calsarcins have been shown to associate the cardiac sarcomeric apparatus with signaling molecules that directly influence gene expression by binding to the Z-disc myofilament anchor proteins α-actinin and telethonin, thereby linking them to calcineurin. This calcium-dependent phosphatase induces cardiomyocyte hypertrophy via downstream transcriptional pathways.[9]
Activation of hypertrophic signaling pathways, such as the mitogen-activated protein kinase, phosphoinositide 3-kinase–protein kinase B, and calcineurin/nuclear factor of activated T cells pathways, leads to gene transcription that promotes growth. Upregulation of fetal genes (eg, atrial natriuretic peptide, brain natriuretic peptide, beta-myosin heavy chain) results in maladaptive remodeling. Altered calcium cycling due to dysfunction of sarcoplasmic reticulum calcium adenosine triphosphatase (sarcoplasmic reticulum calcium adenosine triphosphatase) affects contractility and relaxation.
The cardiac renin-angiotensin system, along with angiotensin-converting enzyme (ACE) function, may play important roles in the hypertrophic response. The fact that myocardial hypertrophy may develop independently of hypertension suggests that angiotensin II may play a role, as it induces myocardial fibrosis as well. Recent studies that performed regression analyses showed that plasma angiotensin II, ACE, and renin levels correlated with left ventricular mass, independent of blood pressure.[7]
Regression analysis also showed that the most important element was angiotensin II levels, which appear to be closely related, in part, to stimulation of myocardial fibrosis. Other factors implicated in the development of myocardial hypertrophy include endothelin, heterotrimeric G proteins, and cardiac sodium-potassium pumps. There may also be evidence of a pro-LVH genotype. Finally, other factors that may have a role in the degree of LVH include concomitant coronary disease or valvular heart disease, as well as inflammatory cytokines, calcium/calmodulin-dependent protein kinase II, and signal transducer and activator of transcription-3.[10]
Genetics
Genomics may also play a significant role in the pathogenesis of LVH. Mutant genes encoding sarcomeric proteins have a direct etiologic role in patients with hypertrophic cardiomyopathy. Also, there seems to be a genetic predisposition, evidenced by the fact that some mildly hypertensive patients develop LVH while others do not.[11]
Histopathology
A heart undergoing hypertrophy usually exhibits changes in architecture and histology, depending on the cause and stage of hypertrophy. Various histologic changes have been demonstrated, including the volume fraction of fibrous tissue, myocyte diameter, and mitochondrial ultrastructure. An increase in the size of individual cells characterizes myocyte hypertrophy.
There is an addition of sarcomeres, and cells have larger diameters. The nucleus becomes enlarged, hyperchromatic, and may show a "boxcar" shape (rectangular appearance), a hallmark of hypertrophic cardiomyopathy (HCM). In concentric hypertrophy, sarcomeres are added in parallel, increasing myocyte thickness, whereas in eccentric hypertrophy, they are added in series, increasing myocyte length.
There is excess production of type I and type III collagen in the interstitial and perivascular spaces. There is also replacement fibrosis, in which fibrosis replaces dead myocytes. Vascular changes include smooth muscle hypertrophy or hyperplasia, leading to thickening of arteriolar walls and reduced capillary density. In HCM, significant myocardial disarray is most prominent in the interventricular septum.
History and Physical
A thorough history and physical examination are essential when evaluating patients with suspected LVH, as they help identify underlying etiologies, associated cardiovascular risk factors, and evidence of end-organ involvement. Clinicians should assess for symptoms suggestive of cardiac dysfunction, including chest pain, dyspnea at rest or with exertion, palpitations, presyncope, syncope, and unexplained fatigue. A detailed cardiovascular risk assessment should include a history of long-standing hypertension and other risk factors for cardiovascular disease, such as metabolic syndrome, tobacco use, elevated low-density lipoprotein cholesterol, diabetes mellitus, obesity, microalbuminuria, physical inactivity, and a family history of premature cardiovascular death.
Family history should also specifically address hypertrophic cardiomyopathy and other inherited cardiomyopathies. Lifestyle factors are important, including participation in high-intensity athletic training or chronic physical exertion, which may result in physiologic LVH, and excessive dietary sodium intake, which may worsen hypertension. Social and substance-use histories should evaluate the use of over-the-counter supplements, herbal products such as licorice or ephedra, and stimulants, including cocaine, amphetamines, and excessive alcohol consumption, all of which may contribute to the development or progression of LVH.
The medical history should assess for chronic kidney disease, endocrine disorders, aortic valve disease, and obstructive sleep apnea. In contrast, the medication history should include adherence to antihypertensive therapy and exposure to anabolic steroids, chemotherapeutic agents, oral contraceptives, and other substances associated with cardiovascular remodeling. In women, a history of gestational hypertension or preeclampsia should be elicited, as both are recognized risk factors for subsequent LVH.
Physical examination may provide important clues regarding both the presence and etiology of LVH. General inspection may reveal obesity, which is often associated with metabolic syndrome and hypertension. Vital signs frequently demonstrate elevated blood pressure, while tachycardia or increased respiratory rate may suggest heart failure. Cardiovascular examination often reveals a sustained, heaving apical impulse resulting from increased left ventricular mass.
The point of maximal impulse may be displaced laterally and inferiorly in the presence of eccentric hypertrophy or left ventricular dilation. An S4 gallop is commonly appreciated due to reduced ventricular compliance, whereas an S3 gallop may indicate concomitant heart failure. A loud first heart sound may be present in patients with hypertension. Specific murmurs may suggest underlying structural heart disease contributing to LVH.
Patients with coarctation of the aorta may exhibit a systolic ejection murmur radiating to the back, accompanied by diminished or absent lower-extremity pulses.[12] Aortic stenosis classically produces a harsh crescendo-decrescendo systolic murmur that peaks in mid-to-late systole and radiates to the carotid arteries, often accompanied by pulsus parvus et tardus. Chronic aortic regurgitation typically manifests as a high-pitched diastolic blowing murmur heard along the left sternal border, with associated findings resulting from a widened pulse pressure, including bounding or “water-hammer” pulses. In patients with long-standing hypertension, funduscopic examination may demonstrate hypertensive retinopathy, characterized by arteriovenous nicking, arteriolar narrowing, cotton-wool spots, flame-shaped hemorrhages, hard exudates, and, in advanced disease, optic disc edema. These findings generally develop later in the course of chronic hypertensive cardiovascular disease.
Evaluation
Electrocardiography
Electrocardiography (ECG) is the least expensive and most readily available test for diagnosing LVH. This condition can be diagnosed in about 1% to 5% of the general population and about one-third of patients with hypertension. The low sensitivity of ECG somewhat limits its clinical utility, but its specificity is relatively high. QRS changes on the ECG include high amplitudes of R waves in leads I, aVL, V5, and V6, which face towards the LV, and deep S waves in the leads that lie on the opposite side of the heart, including V1 and V2. The changes are often associated with R-wave notching or slurring, left axis deviation, and other findings. The J point may also be elevated in leads with tall R waves.
ECG changes result from structural, biochemical, and bioelectric changes. Structural changes include changes or increases in the length and diameter of myocytes, intermyocyte connections, and the number of branches. Biochemical changes can include alterations in ion channels and gap junctions, leading to electrical remodeling. The increase in QRS voltage is generally attributed to an increase in the intensity of current flow across activation fronts. LVH is associated with significant interstitial abnormalities, including inflammation, fibrosis, and degenerative changes, which lead to slower, more fragmented conduction across the myocardium.
ECG is relatively insensitive for diagnosing LVH because it relies on measuring the heart's electrical activity via electrodes placed on the skin to estimate left ventricular mass. The intracardiac electrical signal is difficult to measure in this way because the measurements are affected by all elements between the heart muscle and the ECG electrodes, specifically fat, fluid, and air. Because of the variations in these elements, ECG underdiagnoses LVH in patients with pleural effusions, pericardial effusions, anasarca, obesity, and chronic obstructive pulmonary disease.
Various criteria for LVH by ECG have been suggested over the years. Most criteria utilize the voltage in 1 or more leads, QRS duration, secondary ST-T wave abnormalities, or left atrial abnormalities. Repolarization abnormalities may also be seen in LVH. These include downsloping ST-segment depression and T-wave inversion in leads showing tall R waves (eg, lateral leads: I, aVL, V5, V6).
These changes reflect ventricular strain due to hypertrophy. Left axis deviation is often seen in LVH. Sensitivity and specificity also depend on the criteria used. Matuja et al investigated the real-world use of various ECG diagnostic criteria among young stroke patients in Africa. They found that the Sokolow-Lyon criteria had a specificity of 78% and a sensitivity of 27% for diagnosing LVH, whereas the Cornell standard had a sensitivity of 32% and a specificity of 52%.[13] However, it is also worth noting that LVH standards differ significantly across regions/races.
The best recognized established ECG criteria are as follows:
- Sokolow-Lyon criteria
- S wave in V1 + R wave in V5 ≥35 mV or R wave in aVL ≥11mV
- Cornell voltage criteria
- R wave in aVL + S wave in V3 >28 mV (men) or >20 mV (women)
- Other voltage criteria
- R wave in lead I >14 mm; R wave in lead aVL >11 mm; R wave in lead V5 or V6 >26 mm
- Romhilt-Estes point score system
- A score ≥5 indicates LVH, and 4 suggests possible LVH.[14] The scoring is as follows:
- S wave in V/V2 >/= 3 mV or R wave in V5/V6 ≥3 mV or any limb lead R or S waves >2 mV --> 3 points
- ST-T changes with no digitalis therapy --> 3 points
- ST-T changes with digitalis therapy --> 1 point
- Left atrial abnormality --> 3 points
- Left axis deviation >/= -30 degrees --> 2 points
- QRS duration >/=90 ms --> 1 point
- Delayed intrinsicoid deflection in V5/V6 >/= 50 ms --> 1 point
- A score ≥5 indicates LVH, and 4 suggests possible LVH.[14] The scoring is as follows:
Echocardiogram
An echocardiogram is the test of choice in establishing the diagnosis of LVH. The sensitivity of this modality is significantly higher than ECG, and the test can also diagnose other abnormalities, such as left ventricular dysfunction (both systolic and diastolic) and valvular heart disease. Echocardiography helps differentiate physiological LVH from pathological LVH.
The measurements include the left ventricular end-diastolic diameter (LVEDD), posterior wall thickness (PWT), and interventricular septum thickness (IVS). The left ventricular mass is calculated as 0.8{1.04[([LVEDD + IVSd +PWd]3 - LVEDD3)]} + 0.6. The left ventricular mass index can be calculated as left ventricular mass divided by body surface area. According to the American Society of Echocardiography and the European Association of Cardiovascular Imaging, LVH is defined as an increased left ventricular mass index (LVMI) greater than 95 g/m2 in women and greater than 115 g/m2 in men.[15] The relative wall thickness (RWT) is used to define the type of LVH, ie, eccentric or concentric. The RWT is calculated as (2*PWT)/LVEDD. A normal RWT is ≤0.42. The interpretation is as follows:
- Normal LVMI and normal RWT indicate normal geometry
- Normal LVMI but increased RWT indicates concentric remodeling
- Increased LVMI and increased RWT indicate concentric hypertrophy
- Increased LVMI but normal RWT indicates eccentric hypertrophy
Different echocardiographic patterns of LVH are observed in different conditions. For example, in an athlete's heart, there is eccentric LVH with a maximal wall thickness (MWT) less than 14 mm and balanced dilation of the left and right ventricles. In aortic stenosis, there is concentric LVH and associated findings of reduced cusp excursion and increased transaortic gradients. In hypertensive heart disease, there is asymmetrical hypertrophy predominantly involving the basal IVS.
In HCM, there is an asymmetric septal hypertrophy with MWT greater than 15mm. In cardiac amyloidosis, there is concentric LVH that also involves the right ventricle. Anderson-Fabry disease has severe concentric LVH.[1]
Speckle-tracking echocardiography (STE) helps detect an earlier decrease in global longitudinal strain. Change in global longitudinal strain precedes reduction in ejection fraction. The reduction in global longitudinal strain corresponds to the segment of the left ventricle that is most hypertrophied and/or has the most fibrosis. Longitudinal total stress, evaluated by STE, is sensitive to detecting small, early abnormalities in the myocardium, which can improve prognosis across various diseases compared with traditional echocardiographic indicators; hence, this evaluation has positive diagnostic significance for left ventricular remodeling associated with early hypertension.[16]
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging (CMR) provides an accurate assessment of ventricular dimensions and volumes, left ventricle mass, the distribution of hypertrophy, and tissue characterization.[16] CMR offers the benefit of avoiding ionizing radiation. CMR helps detect early changes of LVH in HCM-gene carriers, such as myocardial crypts, excessive trabeculations, abnormalities in the mitral leaflets, and small left ventricular volumes.
T1 mapping helps characterize changes in intracellular and extracellular tissues, such as edema, fibrosis, and amyloidosis. Extracellular volume mapping quantifies extracellular volume. In HCM, both T1 and ECV are increased.
The pattern of late gadolinium enhancement (LGE) suggests the etiology of LVH. In cardiac amyloidosis, there is diffuse subendocardial LGE; in HCM, LGE occurs mostly in the most hypertrophied segments; in Anderson-Fabry disease, LGE appears in nonischemic midwall or subepicardial segments, particularly the basal inferolateral segments; in Danon disease (a glycogen storage disease), LGE appears extensively in the subendocardium, especially the apex, and characteristically spares the basal septum.[17][18][19]
Novel Diagnostic Biomarkers
Recent transcriptomic studies have identified circulating microRNAs, including et-7c, miR-451, and miR-145-5p, as associated with LVH in hypertensive individuals.[20] In parallel, integrative analyses combining RNA sequencing with machine learning in hypertensive LVH models have highlighted immune-related genes (Ankrd1, Birc5, Nuf2, C1qtnf6, Fcgr3, and Cdca3) that may accurately predict LVH.[21] In addition, immunological profiling studies show a marked reduction in regulatory T cells, including CD4+, CD25+, and Foxp3+, in individuals with hypertensive LVH, along with correlations with cytokines such as interleukin (IL)-6, IL-10, and transforming growth factor beta 1. These could all serve as diagnostic markers in the future.
Treatment / Management
The management of LVH depends on the etiology and focuses on treating the underlying cause, reducing symptoms and workload, and halting progression. The goal is to regress LVH and prevent left ventricular dysfunction and progression to heart failure.
Treatment of the Underlying Cause
Two-thirds of the patients with LVH are hypertensive. Adequate blood pressure control is the cornerstone of managing LVH. Lifestyle modifications such as weight loss, a low-sodium diet, exercise, and smoking cessation help control hypertension. Optimal doses of antihypertensive medications help control afterload and prevent the progression of LVH. ACE inhibitors, angiotensin receptor blockers (ARBs), long-acting calcium channel blockers (CCBs), or thiazide/thiazide-like diuretics are the recommended antihypertensives for LVH. Antihypertensive therapy benefits the patient by reducing BP and may regress LVH independently of blood pressure reduction, leading to reduced adverse cardiovascular events and mortality.[20]
Surgical or transcatheter aortic valve replacement may be required for severe aortic stenosis. Patients with aortic stenosis usually have a 10- to 20-year asymptomatic latent period, during which increasing left ventricular outflow obstruction and pressure load on the myocardium may gradually change the composition of the myocardial extracellular matrix, leading to LVH. Usually, aortic valve replacement is recommended in symptomatic individuals. Still, if the echocardiographic findings show rapidly progressing aortic stenosis with left ventricular dysfunction, aortic valve replacement would be recommended in asymptomatic patients to improve left ventricular function and reduce mortality.
HCM may be treated with a beta-blocker or CCB to control symptoms and afterload. These decrease myocardial contractility, thereby prolonging diastolic filling. Septal reduction therapy, such as surgical myomectomy or alcohol septal ablation, may be needed. An implantable cardioverter defibrillator may be required in patients with HCM to reduce the risk of sudden cardiac death. In these specific cases, drugs like diuretics, ACE inhibitors, or ARBs are avoided because they decrease the preload and worsen the ventricular function.[22] An athletic heart with physiological LVH does not require treatment. Discontinuation of training for a few months (3-6 months) is usually needed to regress LVH. LVH regression is monitored for a few months to distinguish it from cardiomyopathy.
Pharmacologic Therapy
ACE inhibitors (eg, lisinopril, ramipril) and ARBs (eg, losartan) help lower blood pressure, reduce LVH, and improve cardiac function by reducing afterload and preventing further myocardial hypertrophy. Beta-blockers (eg, metoprolol, carvedilol) reduce heart rate and myocardial oxygen demand, which can help alleviate symptoms associated with LVH, particularly in hypertrophic cardiomyopathy or heart failure. Beta-blockers also reduce the risk of arrhythmia. Diuretics (eg, furosemide, spironolactone) help reduce fluid overload, relieve symptoms of heart failure, and lower the workload on the left ventricle in patients with hypertensive heart disease or heart failure. Aldosterone antagonists such as spironolactone or eplerenone can be used, especially in patients with heart failure with preserved ejection fraction or chronic kidney disease, to control fluid retention and lower blood pressure.
Lifestyle Modification
Lifestyle modification is a cornerstone of cardiovascular risk reduction and management of conditions associated with left ventricular hypertrophy. Recommended interventions include adopting a low-sodium, low-fat, high-fiber diet to improve blood pressure control and overall cardiovascular health. Regular physical activity is encouraged to enhance cardiovascular fitness and reduce cardiometabolic risk factors.
Weight loss should be pursued in overweight or obese individuals, as excess body weight contributes to hypertension and adverse cardiac remodeling. Smoking cessation is strongly recommended because tobacco use accelerates cardiovascular disease progression and increases morbidity and mortality. Reduction of alcohol consumption is also advised, as excessive alcohol intake may contribute to hypertension, cardiomyopathy, and worsening cardiovascular outcomes.
Regular Monitoring and Follow-Up
Regular monitoring and follow-up are important components of managing patients with left ventricular hypertrophy and associated cardiovascular conditions. Ambulatory blood pressure monitoring may be utilized to ensure adequate blood pressure control and identify masked or persistent hypertension that may contribute to ongoing cardiac remodeling. Holter monitoring or other ambulatory ECG monitoring is indicated when arrhythmias are suspected, particularly in patients reporting palpitations, syncope, or presyncope. Serial echocardiography should be performed as clinically indicated to assess for progression or regression of LVH, evaluate ventricular size and function, and monitor for the development of associated structural or hemodynamic abnormalities.
Therapeutic Imaging-Based Decision-Making
Echocardiography establishes the presence and severity of left ventricular outflow tract (LVOT) obstruction in patients with LVH due to HCM. A resting or provoked gradient greater than 50 mm Hg requires treatment with nonvasodilating beta-blockers and disopyramide. In patients with HCM, specific echocardiographic features are associated with an increased risk of sudden cardiac death.
These features, combined with clinical and other risk factors, can guide the decision to implant an implantable cardioverter-defibrillator for primary or secondary prevention. These markers include maximal left ventricular wall thickness of 30 mm or more, resting LVOT gradient of 30 mm Hg or greater, or provoked gradient of 50 mm Hg or greater, presence of a localized aneurysm in the left ventricular wall, severe apical hypertrophy, and left ventricular ejection fraction less than 50%. Extensive scarring as defined by LGE on CMR is also a high-risk feature.
Differential Diagnosis
A most challenging clinical dilemma attempts to differentiate between physiological LVH (athlete's heart) and HCM, which is recognized as the most common cause of nontraumatic exercise-induced sudden cardiac death in young athletes (younger than 35 years old). Detailed echocardiographic assessment of left ventricular structure and function helps make the diagnosis.[22]
As outlined above, the differential diagnosis to consider in patients with evidence of LVH includes:
- Essential hypertension
- Renal artery stenosis
- Athletic heart with physiological LVH
- Aortic valvar stenosis
- Coarctation of the aorta
- HCM without or with outflow tract obstruction
- Subaortic stenosis (LVOT by muscle or membrane)
- Aortic regurgitation
- Mitral regurgitation
- Dilated cardiomyopathy
- Ventricular septal defect
- Infiltrative cardiac processes (eg, amyloidosis, Fabry disease, Danon disease)
Pertinent Studies and Ongoing Trials
Several studies have demonstrated that LVH in and of itself is a significant independent risk factor for increased cardiovascular morbidity and mortality and, therefore, a major public health burden, especially in light of an aging population. There is also accumulating evidence that targeting LVH regression is not only feasible but necessary to achieve improved clinical outcomes. Therefore, in treating hypertension and valvular disease, LVH regression should be considered an important surrogate endpoint.
Among the various drugs used to treat hypertension, those that target the renin-angiotensin-aldosterone system, particularly angiotensin receptor blockers (ARBs) and ACE inhibitors, appear to have unique class actions that reduce left ventricular hypertrophy by lowering left ventricular mass, independent of blood pressure control. In addition to treatment with ACE inhibitors and/or ARBs, CCBs and beta-blockers have also been shown to reduce left ventricular mass. This is associated with reduced arrhythmias, mortality, and risk of sudden cardiac death.
The postulation that this decrease in left ventricular mass produces significant improvement in clinical outcomes has yet to be definitively proven. This would indicate that, in the future, the new target for treating left ventricular hypertrophy will require an understanding of an individual’s genomic makeup through new study designs in personalized medicine, as well as advances in genetic technologies, to identify the overall impact on LVH regression. The following are some major trials on LVH:
- The Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study
- LVH regression (diagnosed by ECG utilizing the Sokolow-Lyon index or the Cornell product criteria) in response to losartan improved clinical cardiovascular outcomes independent of blood pressure response.[23]
- There is an association between baseline LVH severity and 5-year risk of clinical events after transcatheter aortic valve replacement (TAVR). Severe LVH had higher 5-year rehospitalization and death rates after TAVR.[24]
- The HOPE (Heart Outcome Prevention Evaluation) trial
- This showed that ramipril use causes regression of ECG markers of LVH and reduces subsequent cardiovascular risk.[25]
- The SPRINT (Systolic Blood Pressure Intervention) trial
- This trial randomized 9193 people with hypertension and LVH to the losartan or atenolol group. On follow-up, there was no difference in mean blood pressure, but patients treated with losartan showed a significant reduction in LVH ECG criteria and the composite of death, myocardial infarction, and stroke. [26]
Prognosis
The presence of LVH forecasts an increased risk of cardiovascular morbidity and mortality, even after adjustment for major cardiovascular risk factors such as age, smoking, obesity, dyslipidemia, blood pressure, and diabetes. This means that LVH is an independent risk factor for cardiovascular disease. Once LVH is developed, it puts the patient at significant risk of developing myocardial ischemia and infarction, heart failure, dysrhythmias, or even sudden death. The risk of cardiovascular disease and adverse major cardiac events increases with increasing left ventricular mass and decreases with the regression of LVH.
Complications
Since continuous pressure or volume overload may remain in a compensatory phase, patients with LVH remain asymptomatic for a few years. But as the disease progresses, it will lead to the development of systolic or diastolic dysfunction and end-stage heart failure. Increased myocardial oxygen demand, eccentric hypertrophy may result in angina or ischemic symptoms. Also, LVH predisposes to arrhythmias because hypertrophied cardiac muscle disrupts normal conduction. This predisposes to atrial fibrillation that may lead to ischemic stroke.
Deterrence and Patient Education
Deterrence of LVH focuses on early identification and aggressive management of modifiable cardiovascular risk factors before structural cardiac remodeling becomes established. Hypertension remains the most important preventable cause of LVH; therefore, maintaining optimal blood pressure control through lifestyle modification and adherence to prescribed antihypertensive therapy is essential. Additional preventive strategies include management of diabetes mellitus, dyslipidemia, obesity, obstructive sleep apnea, and chronic kidney disease. Patients should be encouraged to follow a heart-healthy diet that is low in sodium and saturated fat, engage in regular aerobic physical activity, achieve and maintain a healthy body weight, avoid tobacco use, and limit alcohol consumption. Early evaluation and treatment of valvular heart disease, particularly aortic stenosis and aortic regurgitation, may also help prevent progressive ventricular remodeling and the development of LVH.
Patient education should emphasize that LVH is often a consequence of long-standing cardiovascular stress and may initially be asymptomatic despite increasing risks of heart failure, arrhythmias, myocardial infarction, stroke, and sudden cardiac death. Patients should understand the importance of routine blood pressure monitoring, medication adherence, and regular follow-up visits to assess treatment effectiveness and monitor cardiac function. Education should also include recognition of warning symptoms such as chest pain, exertional dyspnea, palpitations, dizziness, syncope, or worsening exercise intolerance, which may indicate disease progression or associated complications. Empowering patients to actively participate in risk-factor modification, maintain healthy lifestyle habits, and adhere to prescribed therapies can reduce cardiovascular morbidity, promote regression of LVH in some cases, and improve long-term clinical outcomes.
Enhancing Healthcare Team Outcomes
Optimal management of LVH requires coordinated interprofessional collaboration among physicians, advanced practitioners, nurses, pharmacists, cardiologists, dietitians, rehabilitation specialists, and other healthcare professionals to address both the underlying cause and the associated cardiovascular risks. Physicians and advanced practitioners are responsible for identifying LVH, evaluating its etiology, and implementing evidence-based treatment strategies aimed at reducing cardiac remodeling and preventing complications such as heart failure, arrhythmias, myocardial infarction, stroke, and sudden cardiac death. Cardiologists may provide advanced diagnostic evaluation, including echocardiography and cardiac magnetic resonance imaging, particularly when hypertrophic cardiomyopathy or other structural heart disease is suspected.
Nurses play a vital role in blood pressure monitoring, patient education, symptom assessment, reinforcement of medication adherence, and identification of clinical deterioration. Pharmacists contribute through medication reconciliation, optimization of antihypertensive therapy, management of adverse drug effects, and patient counseling regarding medication compliance and cardiovascular risk reduction. Dietitians support the implementation of low-sodium, heart-healthy dietary plans, while exercise specialists and cardiac rehabilitation professionals assist patients in developing safe and effective physical activity programs.
Effective communication and care coordination are essential because LVH is often a manifestation of chronic conditions such as hypertension, valvular heart disease, obesity, diabetes mellitus, chronic kidney disease, and obstructive sleep apnea. Shared decision-making, standardized follow-up protocols, and regular communication among primary care clinicians, cardiologists, nurses, pharmacists, and ancillary healthcare professionals facilitate timely adjustment of treatment plans and improve long-term disease management. Coordinated monitoring through ambulatory blood pressure assessment, laboratory testing, electrocardiography, and serial echocardiography helps evaluate therapeutic response and detect complications early. By promoting patient education, lifestyle modification, medication adherence, and comprehensive risk-factor control, interprofessional teams can improve patient-centered care, enhance patient safety, reduce cardiovascular morbidity and mortality, and optimize long-term outcomes in individuals with left ventricular hypertrophy.
Media
(Click Image to Enlarge)
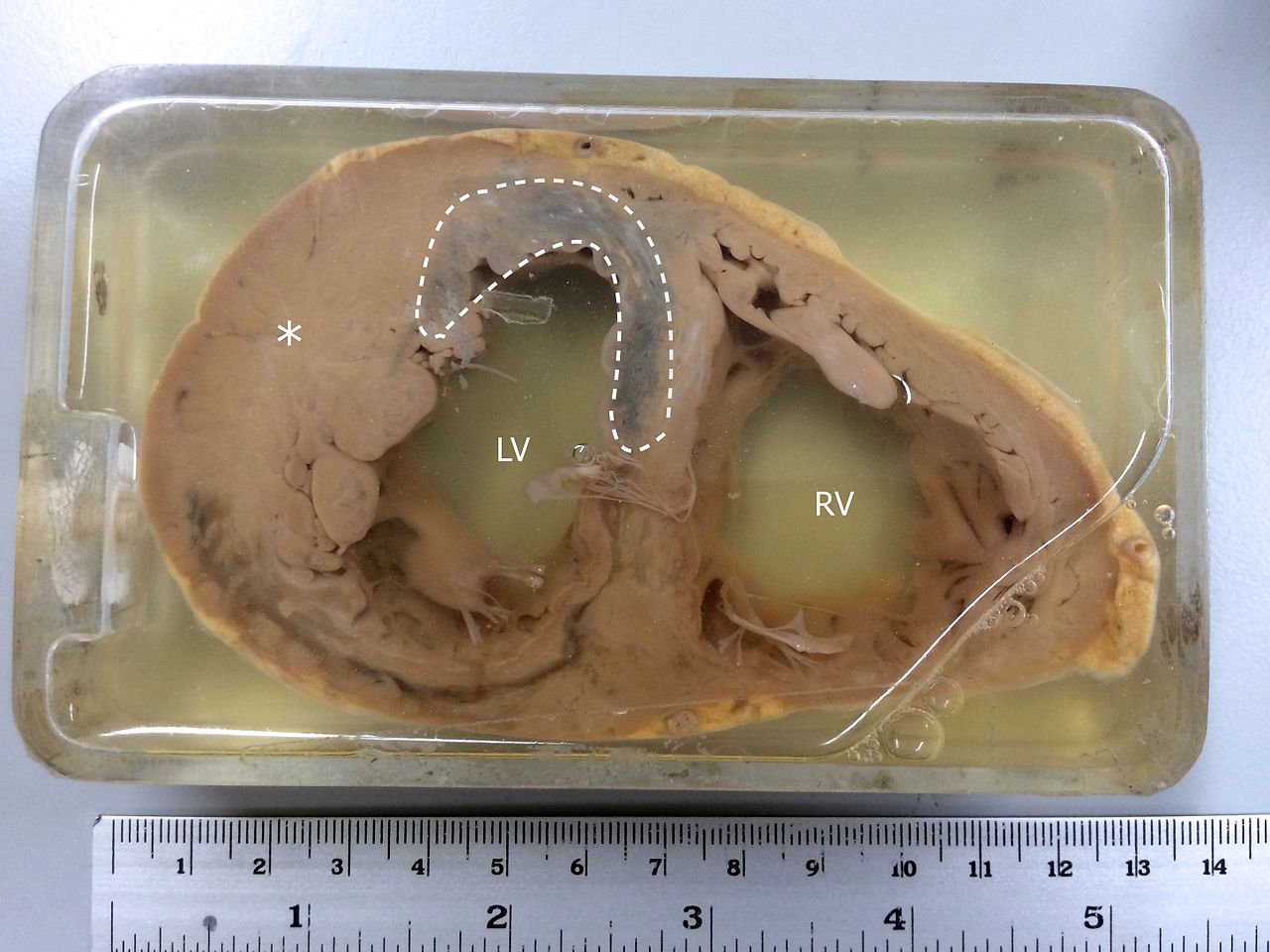
Specimen Showing Myocardial Infarction. MI is observed in the left ventricle and the interventricular septum. The asterisk(*) indicates left ventricular hypertrophy.
Contributed by Wikimedia Commons (CC by 4.0) https://creativecommons.org/licenses/by/4.0/
References
Moura B, Aimo A, Al-Mohammad A, Keramida K, Ben Gal T, Dorbala S, Todiere G, Cameli M, Barison A, Bayes-Genis A, von Bardeleben RS, Bucciarelli-Ducci C, Delgado V, Mordi IR, Seferovic P, Savarese G, Čelutkienė J, Rapezzi C, Emdin M, Coats A, Metra M, Rosano G. Diagnosis and management of patients with left ventricular hypertrophy: Role of multimodality cardiac imaging. A scientific statement of the Heart Failure Association of the European Society of Cardiology. European journal of heart failure. 2023 Sep:25(9):1493-1506. doi: 10.1002/ejhf.2997. Epub 2023 Sep 4 [PubMed PMID: 37581253]
Yildiz M, Oktay AA, Stewart MH, Milani RV, Ventura HO, Lavie CJ. Left ventricular hypertrophy and hypertension. Progress in cardiovascular diseases. 2020 Jan-Feb:63(1):10-21. doi: 10.1016/j.pcad.2019.11.009. Epub 2019 Nov 21 [PubMed PMID: 31759953]
Leache L, Gutiérrez-Valencia M, Finizola RM, Infante E, Finizola B, Pardo Pardo J, Flores Y, Granero R, Arai KJ. Pharmacotherapy for hypertension-induced left ventricular hypertrophy. The Cochrane database of systematic reviews. 2021 Oct 10:10(10):CD012039. doi: 10.1002/14651858.CD012039.pub3. Epub 2021 Oct 10 [PubMed PMID: 34628642]
Level 1 (high-level) evidenceCuspidi C, Sala C, Negri F, Mancia G, Morganti A, Italian Society of Hypertension. Prevalence of left-ventricular hypertrophy in hypertension: an updated review of echocardiographic studies. Journal of human hypertension. 2012 Jun:26(6):343-9. doi: 10.1038/jhh.2011.104. Epub 2011 Nov 24 [PubMed PMID: 22113443]
Sayin BY, Oto A. Left Ventricular Hypertrophy: Etiology-Based Therapeutic Options. Cardiology and therapy. 2022 Jun:11(2):203-230. doi: 10.1007/s40119-022-00260-y. Epub 2022 Mar 30 [PubMed PMID: 35353354]
Marketou ME, Parthenakis F, Vardas PE. Pathological Left Ventricular Hypertrophy and Stem Cells: Current Evidence and New Perspectives. Stem cells international. 2016:2016():5720758. doi: 10.1155/2016/5720758. Epub 2015 Dec 20 [PubMed PMID: 26798360]
Level 3 (low-level) evidenceJia G, Aroor AR, Hill MA, Sowers JR. Role of Renin-Angiotensin-Aldosterone System Activation in Promoting Cardiovascular Fibrosis and Stiffness. Hypertension (Dallas, Tex. : 1979). 2018 Sep:72(3):537-548. doi: 10.1161/HYPERTENSIONAHA.118.11065. Epub [PubMed PMID: 29987104]
Sciarretta S, Paneni F, Palano F, Chin D, Tocci G, Rubattu S, Volpe M. Role of the renin-angiotensin-aldosterone system and inflammatory processes in the development and progression of diastolic dysfunction. Clinical science (London, England : 1979). 2009 Mar:116(6):467-77. doi: 10.1042/CS20080390. Epub [PubMed PMID: 19200056]
Samak M, Fatullayev J, Sabashnikov A, Zeriouh M, Schmack B, Farag M, Popov AF, Dohmen PM, Choi YH, Wahlers T, Weymann A. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Medical science monitor basic research. 2016 Jul 23:22():75-9. doi: 10.12659/MSMBR.900437. Epub 2016 Jul 23 [PubMed PMID: 27450399]
Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nature reviews. Molecular cell biology. 2013 Jan:14(1):38-48. doi: 10.1038/nrm3495. Epub [PubMed PMID: 23258295]
Level 3 (low-level) evidenceMarian AJ, Braunwald E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circulation research. 2017 Sep 15:121(7):749-770. doi: 10.1161/CIRCRESAHA.117.311059. Epub [PubMed PMID: 28912181]
Agabiti-Rosei E, Muiesan ML, Salvetti M. Evaluation of subclinical target organ damage for risk assessment and treatment in the hypertensive patients: left ventricular hypertrophy. Journal of the American Society of Nephrology : JASN. 2006 Apr:17(4 Suppl 2):S104-8 [PubMed PMID: 16565230]
Matuja SS, Munseri P, Moshiro C, Khanbhai K, Mahawish K. The burden, correlates and outcomes of left ventricular hypertrophy among young Africans with first ever stroke in Tanzania. BMC cardiovascular disorders. 2021 Oct 9:21(1):485. doi: 10.1186/s12872-021-02297-8. Epub 2021 Oct 9 [PubMed PMID: 34627161]
Romhilt DW, Estes EH Jr. A point-score system for the ECG diagnosis of left ventricular hypertrophy. American heart journal. 1968 Jun:75(6):752-8 [PubMed PMID: 4231231]
Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2015 Jan:28(1):1-39.e14. doi: 10.1016/j.echo.2014.10.003. Epub [PubMed PMID: 25559473]
Barison A, Aimo A, Todiere G, Grigoratos C, Aquaro GD, Emdin M. Cardiovascular magnetic resonance for the diagnosis and management of heart failure with preserved ejection fraction. Heart failure reviews. 2022 Jan:27(1):191-205. doi: 10.1007/s10741-020-09998-w. Epub [PubMed PMID: 32572736]
Dorbala S, Cuddy S, Falk RH. How to Image Cardiac Amyloidosis: A Practical Approach. JACC. Cardiovascular imaging. 2020 Jun:13(6):1368-1383. doi: 10.1016/j.jcmg.2019.07.015. Epub 2019 Oct 11 [PubMed PMID: 31607664]
Wei X, Zhao L, Xie J, Liu Y, Du Z, Zhong X, Ye W, Wang Y, Chen Y, Lu M, Liu H. Cardiac Phenotype Characterization at MRI in Patients with Danon Disease: A Retrospective Multicenter Case Series. Radiology. 2021 May:299(2):303-310. doi: 10.1148/radiol.2021203996. Epub 2021 Mar 23 [PubMed PMID: 33754825]
Level 2 (mid-level) evidenceRuilope LM, Schmieder RE. Left ventricular hypertrophy and clinical outcomes in hypertensive patients. American journal of hypertension. 2008 May:21(5):500-8. doi: 10.1038/ajh.2008.16. Epub 2008 Mar 13 [PubMed PMID: 18437140]
Level 2 (mid-level) evidenceWilliams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, Clement DL, Coca A, de Simone G, Dominiczak A, Kahan T, Mahfoud F, Redon J, Ruilope L, Zanchetti A, Kerins M, Kjeldsen SE, Kreutz R, Laurent S, Lip GYH, McManus R, Narkiewicz K, Ruschitzka F, Schmieder RE, Shlyakhto E, Tsioufis C, Aboyans V, Desormais I, ESC Scientific Document Group. 2018 ESC/ESH Guidelines for the management of arterial hypertension. European heart journal. 2018 Sep 1:39(33):3021-3104. doi: 10.1093/eurheartj/ehy339. Epub [PubMed PMID: 30165516]
Zhou M, Li T, Lv S, Gan W, Zhang F, Che Y, Yang L, Hou Y, Yan Z, Zeng Z, Zhao W, Yang M. Identification of immune-related genes and small-molecule drugs in hypertension-induced left ventricular hypertrophy based on machine learning algorithms and molecular docking. Frontiers in immunology. 2024:15():1351945. doi: 10.3389/fimmu.2024.1351945. Epub 2024 Jun 27 [PubMed PMID: 38994368]
Malhotra A, Sharma S. Hypertrophic Cardiomyopathy in Athletes. European cardiology. 2017 Dec:12(2):80-82. doi: 10.15420/ecr.2017:12:1. Epub [PubMed PMID: 30416558]
Dahlöf B, Devereux R, de Faire U, Fyhrquist F, Hedner T, Ibsen H, Julius S, Kjeldsen S, Kristianson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. The Losartan Intervention For Endpoint reduction (LIFE) in Hypertension study: rationale, design, and methods. The LIFE Study Group. American journal of hypertension. 1997 Jul:10(7 Pt 1):705-13 [PubMed PMID: 9234823]
Gonzales H, Douglas PS, Pibarot P, Hahn RT, Khalique OK, Jaber WA, Cremer P, Weissman NJ, Asch FM, Zhang Y, Gertz ZM, Elmariah S, Clavel MA, Thourani VH, Daubert M, Alu MC, Leon MB, Lindman BR. Left Ventricular Hypertrophy and Clinical Outcomes Over 5 Years After TAVR: An Analysis of the PARTNER Trials and Registries. JACC. Cardiovascular interventions. 2020 Jun 8:13(11):1329-1339. doi: 10.1016/j.jcin.2020.03.011. Epub [PubMed PMID: 32499024]
Mathew J, Sleight P, Lonn E, Johnstone D, Pogue J, Yi Q, Bosch J, Sussex B, Probstfield J, Yusuf S, Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation. 2001 Oct 2:104(14):1615-21 [PubMed PMID: 11581138]
Okin PM, Wachtell K, Devereux RB, Harris KE, Jern S, Kjeldsen SE, Julius S, Lindholm LH, Nieminen MS, Edelman JM, Hille DA, Dahlöf B. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA. 2006 Sep 13:296(10):1242-8 [PubMed PMID: 16968848]