 Clinical Significance of Volume of Distribution in Pharmacotherapy
Clinical Significance of Volume of Distribution in Pharmacotherapy
Introduction
The volume of distribution (Vd) is a pharmacokinetic parameter that describes a drug's tendency to remain in the plasma or distribute into other tissue compartments. By definition, Vd is a proportionality constant that relates the total amount of drug in the body to the plasma concentration of the drug at a given time.[1][2][3]
Volume of distribution is represented as: Vd=Amount of drug in the body/Plasma concentration of the drug.
The unit of Vd is liter (L). In this relationship, the amount of drug in the body is typically expressed in milligrams (mg), and the plasma drug concentration is expressed in mg/L.
Vd is a theoretical or apparent volume of fluid that a drug would need to occupy to achieve the observed plasma concentration. Traditionally, Vd is described in terms of the relationship between the drug’s plasma concentration and the total amount of drug in the body. Vd can vary depending on the dosing regimen and the timing of measurement.[3][4][5] Notably, Vd does not represent a true physical or physiological volume. Instead, it is an apparent or theoretical volume required to contain the total amount of drug in the body at the same concentration as measured in plasma.[6]
Table 1. Types of Volume of Distribution
| Type | Distribution |
| Vc | Initial Vd, representing the volume of the central compartment before the drug distributes into extravascular tissues |
| Varea/Vz/Vβ | Vd is calculated using the area method during the beta (elimination) phase |
| Vss | Vd at steady state, when plasma drug concentration is measured at Css |
Abbreviations: Css=steady-state conditions; Vd=volume of distribution.
A drug with a high Vd tends to leave the plasma and distribute extensively into extravascular compartments. Consequently, a higher dose may be required to achieve a given plasma concentration. (High Vd → Greater distribution into other tissues.)
In contrast, a drug with a low Vd tends to remain in plasma, implying that a lower dose is required to achieve a given plasma concentration. (Low Vd → Less distribution into other tissues.)
The volume of distribution is commonly determined using the drug concentration measured in plasma or serum, which are generally considered equivalent for this purpose.[7] Drugs can have variable Vd (eg, warfarin has a very low Vd [0.14 L/kg], whereas chloroquine has a high Vd [235 L/kg]).[1]
Vd is commonly expressed in liters (L). However, it is often normalized to an individual’s body weight by dividing by body weight in kilograms (kg), resulting in a standardized unit of liters per kilogram (L/kg).
Table 2. Comparison of Drugs With Low Versus High Volume of Distribution and Their Clinical Implications
|
Clinical Parameter |
Low Vd Drugs (≤0.7 L/kg) |
High Vd Drugs (≥5 L/kg) |
|
Representative drugs |
Warfarin (0.14 L/kg; ~10 L), gentamicin (0.25 L/kg; ~18 L), vancomycin (0.4-1.0 L/kg; ~28-70 L), theophylline (0.45 L/kg; ~32 L), lithium (0.7-1.0 L/kg; ~49-70 L), phenytoin (0.6-0.7 L/kg; ~42-49 L). |
Amiodarone (66 L/kg; ~4620 L), chloroquine (235 L/kg: ~14,000-56,000 L), digoxin (7.0 L/kg; ~490 L), fluoxetine (12-43 L/kg; ~840-3010 L), propofol (2-10 L/kg; ~140-700 L), buprenorphine (188-335 L following IV administration).[8] |
|
Primary distribution |
Predominantly in plasma and/or ECF. |
Extensively distributed into tissues (adipose, muscle, liver, and lung). |
|
Loading dose implication |
Smaller LD needed (LD=Cp×Vd); clinically feasible to achieve rapid therapeutic levels. |
Larger LD required; may necessitate prolonged loading regimens (eg, oral loading of amiodarone over several weeks). |
|
Dialyzability (KDIGO/EXTRIP) |
More effectively removed by hemodialysis when PPB is also low (eg, gentamicin and lithium); supplemental dosing after hemodialysis is recommended.[9] |
Poorly removed by hemodialysis or CRRT because the vast majority of the drug resides in tissues, not in plasma (eg, amiodarone and digoxin). |
|
TDM and toxicity pattern |
Plasma concentrations correlate well with pharmacologic effect; TDM is clinically useful and helps guide dose adjustments. |
Tissue accumulation primarily drives pharmacologic effects; plasma levels may not fully reflect tissue burden; toxicity may persist long after discontinuation. |
|
Effect of fluid overload (AKI/sepsis) |
Vd increases significantly with third-spacing/edema; higher LDs of hydrophilic drugs may be needed.[10] |
Vd is already large; fluid overload has a proportionally smaller impact on total Vd; LD adjustment is less critical. |
|
Obesity dosing consideration |
IBW or ABW should be used for hydrophilic drugs; adipose tissue has minimal effect on distribution.[11] |
Lipophilic drugs may exhibit an increased Vd in individuals with obesity. In such cases, use total body weight or ABW); eg, propofol and amiodarone.[12] |
|
Beers Criteria Relevance (AGS 2023) |
Gentamicin should be used with caution in geriatric patients due to the risk of nephrotoxicity and ototoxicity. For phenytoin, monitoring of free (unbound) levels is recommended due to age-related hypoalbuminemia.[13] |
Amiodarone is listed as a potentially inappropriate medication in the AGS Beers criteria due to the risk of thyroid, pulmonary, and cardiac toxicity in older adults; thus, it should be avoided unless no alternative exists.[13] However, life-threatening arrhythmias should not be a contraindication for amiodarone use. |
Abbreviations: ABW=adjusted body weight; AGS=American Geriatrics Society; AKI=acute kidney injury; Cp=target plasma concentration; ECF=extracellular fluid; EXTRIP=Extracorporeal Treatments in Poisoning; IBW=ideal body weight; IV=intravenous; KDIGO=Kidney Disease: Improving Global Outcomes; LD=loading dose; PPB=plasma protein binding; TDM=therapeutic drug monitoring; Vd=volume of distribution.
Function
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Function
General Principles Related to Drug Distribution
Pharmacokinetics governs the movement of drugs throughout the human body during absorption, distribution, metabolism, and excretion. After administration, a drug moves from the site of administration into the systemic circulation, where it gets distributed throughout the body. Drug distribution refers to the movement of a drug between the intravascular (blood or plasma) and extravascular (intracellular and extracellular) compartments of the body.
Within each body compartment, drugs exist in equilibrium between protein-bound and free (unbound) forms. Over time, circulating drugs are metabolized primarily in the liver and eliminated mainly through the kidneys.[1][3] Because plasma drug concentrations can be measured at multiple time points (such as immediately after administration, at steady state, and during elimination), different values of Vd can be calculated. Additionally, Vd can be calculated using multiple other equations; the most basic ones are explained in the article.
Single- Versus Multi-Compartment Models of Volume of Distribution
Immediately after administration of an intravenous (IV) bolus, a drug enters the central compartment, which includes plasma and highly perfused organs (such as liver, kidneys, and lungs), where distribution is instantaneous. Over time, the drug may distribute from the central compartment to the peripheral compartment, which is largely composed of other body tissues (see Image. Single- Versus Multi-Compartment Model of Distribution).[1][2][3][14]
Single-compartment model: Some drugs exhibit pharmacokinetics in which distribution throughout the body appears to occur instantaneously. In these cases, the drug behaves as if it remains within the central compartment and does not distribute into peripheral compartments. Therefore, any measured decline in plasma drug concentration primarily reflects drug elimination from the body. Such drugs are described by a single-compartment distribution model.
For these drugs, the Vd can be expressed as the volume of the central compartment (Vc),[2][3] and is represented as:
Vc=Amount of drug dose administered/initial plasma concentration of drug just after IV administration.
In clinical practice, Vc can be used to estimate the initial maximum concentration for an IV bolus (eg, with certain anesthetic medications) or to determine plasma volume using markers such as Evans blue dye, which remains in the plasma or central compartment only.[3]
Drugs that exhibit single-compartment distribution kinetics typically follow a straight-line graph on plasma concentration versus time curves. Because the drug distributes instantaneously, its initial plasma concentration is difficult to measure directly. Instead, it is estimated by extrapolating the plasma concentration–time curve back to time zero.[1][2][3][14]
Multi-compartment model: Most drugs exhibit slower distribution kinetics, characterized by an early distribution phase followed by a later elimination phase. In practice, the majority of drugs follow a multi-compartment model of distribution. In this model, drugs first distribute from the central compartment to peripheral compartments before undergoing elimination.[1][2][3][5][15]
Different phases are described in a multi-compartment distribution model, as outlined below.
- Distribution phase: Following drug administration, the plasma drug concentration initially declines while the total amount of drug in the body remains the same. As a result, a single drug may have multiple Vd values that vary depending on the time of measurement.
- Elimination phase: After the distribution phase is complete, the drug is eliminated from the central compartment (by the liver and kidneys), leading to changes in both the amount of the drug in the body and the plasma drug concentration. At this time, the drug can re-enter the plasma from tissues. Another value of Vd can therefore be calculated during the terminal elimination phase. In the literature, this parameter is referred to as Varea, Vz, or Vβ. The Vd (Varea/Vz/Vβ) value will generally exceed Vc. Varea is defined as clearance divided by the terminal elimination rate constant and is therefore directly dependent on the terminal rate of elimination.
- Varea/Vz/Vβ= Amount of drug in the body during the terminal phase/Plasma concentration of drug during the terminal phase.
- Steady-state phase: Between the distribution and elimination phases, there is a transition phase referred to as steady state. Steady state represents a period of dynamic equilibrium in which the drug has completed distribution between the central and peripheral compartments. At this stage, there is no net movement of the drug between these compartments. Hence, another value of Vd can be calculated in steady state, denoted as Vss. Vss is the most clinically relevant Vd parameter for determining the loading dose of a drug. Vss is considered a more accurate measure of the total body distribution volume, and its value usually lies between Vc and Varea.
- Vss=Amount of drug in the body during equilibrium/Plasma concentration of drug at steady state.
Drugs that exhibit multiple-compartment distribution kinetics typically exhibit biphasic plasma concentration–time curves.[16]
Half-Life and Volume of Distribution
Half-life (t1/2) refers to the time required for the plasma concentration of a drug to decrease by 50%. Half-life is related to Vd and drug clearance (CL).[1][2][3][17]
Half-life can be expressed using the following equation:
t1/2=0.693×Vd/CL, where
- t1/2: Half-life (time, expressed in hours)
- Vd: Volume of distribution (in liters)
- CL: Clearance (expressed as volume per time, L/h)
Only the drug present in the central compartment (plasma and highly perfused organs) can be eliminated from the body, as elimination primarily occurs through the liver and kidneys. Drugs with a high Vd have a significant fraction of the drug stored in the peripheral compartment. As the fraction of the drug in the plasma is eliminated, the equilibrium shifts, allowing the drug from the peripheral compartments to redistribute into the central compartment. This redistribution helps maintain plasma drug concentrations despite ongoing drug elimination from the body. This phenomenon causes plasma concentration to decline more slowly during the elimination phase in the setting of a high Vd. This model has been studied across a wide variety of drugs but is most commonly applied to anesthetic agents, often described as a redistribution model of pharmacokinetic behaviour.[1][3][18][19] Hence, at a constant rate of clearance, a drug with a high Vd will have a longer elimination half-life than a drug with a lower Vd.
Similar to the different Vd values that exist depending on the pharmacokinetic phase, 2 half-life values can also be described:[16]
- Distribution half-life (t1/2α): The time required for the plasma concentration to decrease by 50% during the distribution phase.
- Elimination half-life (t1/2β): The time required for the plasma concentration to decrease by 50% during the elimination phase.
Major Factors Affecting the Volume of Distribution
Plasma protein binding: Drugs can bind to plasma proteins, such as albumin, alpha-1 acid glycoprotein, and lipoproteins, which are significant determinants of the Vd. Drugs with high plasma protein binding (PPB) tend to remain in the plasma (central) compartment and therefore have smaller volumes of distribution than drugs with lower protein binding. The extent of protein binding can be influenced by the acid-base properties and the lipophilicity (hydrophobicity) of the drug.[6][20]
- Acid-base characteristics: Drugs tend to bind proteins throughout the body, reaching an equilibrium between bound and unbound phases. Depending on the charge of a drug at physiological pH, a drug may preferentially bind to macromolecules inside or outside the plasma.[2]
- Basic (alkaline) drugs tend to interact strongly with negatively charged phospholipid head groups located on phospholipid membranes. The extent of this binding also depends on the drug's overall lipophilicity. In general, basic molecules are less likely to leave the systemic circulation, leading to a higher Vd than acidic molecules. Basic drugs also bind predominantly to alpha-1 acid glycoprotein.
- In contrast, acidic molecules have a higher affinity for albumin, particularly when their lipophilicity is relatively lower than neutral or basic molecules. Therefore, acidic drugs are more likely to bind to albumin and remain bound within the plasma compartment, resulting in a lower Vd than more basic molecules.
- Lipophilicity: Drugs with higher lipophilicity have greater lipid membrane permeability and therefore have a higher likelihood of leaving the plasma and interacting with other hydrophobic residues in the peripheral tissues (eg, adipose tissues). However, plasma proteins, such as albumin, also have a high affinity for lipophilic drugs. In this case, the determinant of the extent of PPB of 2 equally lipophilic drugs is the acid-base characteristics as described above.[2] In general, the following rules apply:
- Lipophilic molecules are more likely to pass through lipid bilayers and, therefore, are more likely to leave the bloodstream and distribute to areas with high lipid density (eg, adipose tissues, brain, and kidneys), resulting in a higher Vd.[21]
- Hydrophilic molecules are less likely to pass through lipid bilayers and, therefore, more likely to remain in the bloodstream, resulting in a lower Vd.
Drug transporters: Drug transporters are present in the body tissues and are considered essential factors in drug distribution. The liver and kidneys are the primary organs that express a variety of drug transporters. These transporters regulate access to metabolizing enzymes and excretion processes, thereby influencing drug clearance and half-life (t1/2).
Transporter activity may be altered by drug–drug interactions or genetic polymorphisms in transporter genes or relevant genetic control elements. Such changes can affect drug distribution, potentially altering the Vd, drug effect, and half-life. The primary site of transporter activity, particularly in the liver or kidneys, can be a major predictor of changes in Vd.[5]
Drug-Drug Interactions
Drug-drug interactions can significantly alter Vd through several pharmacokinetic mechanisms. Understanding how drug–drug interactions affect drug distribution is essential for safe prescribing and dose optimization, particularly in patients receiving polypharmacy.[5][14]
Protein-binding displacement interactions: Displacement of a highly protein-bound drug from its PPB sites transiently increases the free (unbound) fraction of the drug. This increase may raise the apparent Vd and potentially lead to toxicity before a new steady-state equilibrium is established. For example, valproic acid can displace phenytoin from albumin-binding sites, increasing free phenytoin concentrations. In such cases, total phenytoin levels may appear within the normal range while free concentrations reach toxic levels. Clinical pharmacokinetic literature and guidance from the American College of Clinical Pharmacy (ACCP) recommend monitoring free phenytoin concentrations when a displacing agent is coadministered.
Similarly, sulfonamides and nonsteroidal anti-inflammatory drugs may displace warfarin from albumin-binding sites. However, the clinical impact is often transient because the increase in free warfarin also leads to faster clearance.[22] The net clinical effect of PPB displacement depends on whether the displaced drug undergoes restrictive clearance (low hepatic extraction ratio) or nonrestrictive clearance (high hepatic extraction ratio).[23][15]
Transporter-mediated interactions affecting distribution: Drug transporters such as P-glycoprotein (P-gp) and other efflux or uptake transporters play an important role in regulating drug distribution between plasma and tissue compartments. Inhibition of P-gp (eg, by amiodarone, verapamil, quinidine, or cyclosporine) can reduce drug efflux from tissues and alter the distribution of P-gp substrates such as digoxin.
When amiodarone is coadministered with digoxin, serum digoxin concentrations may increase by approximately 70% to 100% because of P-gp inhibition in the kidneys and intestines. This interaction can reduce renal clearance and alter digoxin Vd. Therefore, an empirical reduction of the digoxin dose by about 50% is commonly recommended when initiating amiodarone therapy. The DailyMed labeling for both drugs highlights this interaction.[24] Conversely, P-gp inducers, such as rifampin and St John’s wort, can increase tissue distribution while lowering plasma concentrations of P-gp substrates, potentially reducing therapeutic efficacy.[5]
CYP450-mediated interactions and indirect effects on distribution: Although cytochrome P450 (CYP450) interactions primarily affect drug metabolism and clearance, they can indirectly influence drug distribution at steady state. Because half-life (t1/2) depends on both the Vd and clearance (t1/2=0.693×Vd/CL), inhibition of CYP enzymes decreases clearance, which prolongs half-life and may increase overall drug accumulation in tissues.
Amiodarone is a potent inhibitor of CYP3A4, CYP2C9, and CYP2D6. When this drug is coadministered with CYP substrates such as warfarin (S-enantiomer via CYP2C9), phenytoin (CYP2C9/2C19), simvastatin (CYP3A4), and fentanyl (CYP3A4), dose adjustments are often required.[25] The US Food and Drug Administration (FDA)–approved labeling for amiodarone recommends reducing the warfarin dose by approximately 33% to 50% and closely monitoring the international normalized ratio (INR) when initiating amiodarone therapy.[26][27]
Clinical Significance
Although multiple values of Vd can be calculated depending on the compartment model (single- versus multi-compartment) and the pharmacokinetic phase of the drug following drug administration (distribution phase, steady state, or terminal elimination phase), from the clinical perspective, the single most important utility of Vd is the calculation of the loading dose of a drug.[1][3]
The loading dose is best calculated using the Vd value at steady state (Vss), as it reflects the drug's pharmacokinetic properties at the desired steady-state plasma concentration. The loading dose (mg) can be calculated using the following equation:
- Loading dose=(Cp×Vd)/F
- Cp: Desired plasma concentration of drug (mg/L)
- Vd: Volume of distribution (L), ideally Vss
- F: Bioavailability of drug (IV administration=1; oral administration ranges from 0 to 1)
After a loading dose is administered, additional maintenance doses may be required to sustain the desired therapeutic plasma concentration of the drug. While the loading dose primarily depends on the Vd of the drug, the maintenance dose depends on the drug's clearance.[3]
The maintenance dose rate (mg/h) can be calculated using the following equation:
- Maintenance dose rate=(Cp×CL)/F
- Cp: Desired plasma concentration of the drug (mg/L)
- CL: Clearance rate of the drug (L/h)
- F: Bioavailability of the drug (IV administration=1; oral administration ranges from 0 to 1)
Major differences between loading and maintenance doses include:
- Loading doses primarily depend on Vd, whereas maintenance doses depend on plasma clearance.[3]
- A loading dose is required for certain drugs in specific clinical scenarios when rapid attainment of steady state is needed (eg, antiepileptic drugs during an active seizure or anesthetic agents such as propofol and dexmedetomidine administered before continuous infusion). On the other hand, maintenance doses are required for most drugs to maintain a therapeutic plasma concentration at steady state.[3][28][29]
- Loading doses rarely require modification, whereas maintenance doses require adjustment based on patient-specific characteristics.[3] Maintenance doses depend on the drug's clearance rate, which can vary among individuals, as patients may take longer or shorter to clear the drug from plasma. For example, patients with renal failure have reduced kidney function and therefore require a longer time to eliminate drugs that are primarily excreted by the kidneys. In such cases, the loading dose typically remains unchanged, whereas the maintenance dose must be adjusted, either by reducing the dose administered per unit time or by extending the dosing interval.
In addition to PPB and drug transporters, other factors can alter the apparent Vd. Due to these interactions, the Vd of certain drugs may vary among patients depending on their pathophysiology; a few examples are outlined below.
- Pediatric versus adult dosing: Body composition changes with advancing age, which can affect drug distribution; therefore, loading doses may differ between pediatric and adult patients.[30][31]
- Normal body mass index versus higher body mass index (obesity): The loading doses of certain drugs, such as anesthetics, may be calculated using different weight-scaling factors, such as total body weight versus ideal body weight, depending on the pharmacokinetics of specific drugs to prevent overdosing or underdosing.[12][32] For patients receiving leveterictiam, body weight significantly influenced Vd, whereas renal function influenced clearance.[33][34]
- Plasma protein concentration: Excess or deficiency of plasma proteins (eg, albumin) may alter PPB, thereby affecting the amount of drug retained in the plasma and, consequently, the apparent Vd.[1][15][35]
The concept of Vd is essential for both physicians and pharmacologists who prescribe and review medication doses. Differentiating pharmacologic agents with high versus low volumes of distribution is vital for appropriate medication dosing in patients.
A low Vd indicates that the drug is largely confined to the plasma or extracellular fluid (<0.6 L/kg), whereas a high Vd indicates extensive distribution into body tissues, fat, or muscle (>5 L/kg).
In routine clinical situations, physicians typically prescribe medication doses without complex adjustments. However, critically ill patients, particularly those in the intensive care unit or receiving anesthesia, may require individualized dose adjustments by a clinical pharmacist. Understanding pharmacokinetic distribution models and the factors that influence Vd, loading doses, and maintenance doses is important for optimizing drug therapy in these patients.[36][37] In such settings, consultation with an interprofessional team experienced in medication dosing is essential to ensure safe and effective treatment.
Hemodialysis induces rapid shifts in intravascular volume and changes in PPB, which can temporarily affect the apparent Vd of antipsychotics. This may lead to transient increases or decreases in measured plasma levels without corresponding changes in the body's overall drug load. As a result, Vd-driven redistribution during hemodialysis, combined with drug-specific protein binding and dialyzability, reduces the accuracy of dosing recommendations based on chronic renal failure data. This underscores the importance of considering Vd, protein binding, and timing relative to hemodialysis.[38]
In heart failure, reduced cardiac output and impaired organ perfusion can decrease the Vd of drugs that normally distribute into poorly perfused tissues such as skeletal muscle and adipose tissue. For example, the Vd of antiarrhythmic agents such as lidocaine and procainamide may decrease by up to 40%, which may require lower loading doses to avoid toxicity.[10][39] In a large randomized controlled trial of a Vd-dependent drug in heart failure, digoxin (Vd≈7.0 L/kg, approximately 490 L) was demonstrated to reduce heart failure–related hospitalizations by about 28% but did not show a mortality benefit when serum concentrations were above 1.0 ng/mL. Post hoc analyses confirmed that target serum digoxin concentrations of 0.5 to 0.9 ng/mL were associated with improved outcomes, suggesting that reduced Vd and impaired renal clearance in heart failure may require lower dosing.[40]
Randomized controlled trials have also shown that neurohormonal blockade improves hemodynamics in heart failure and may partially normalize Vd fluctuations as cardiac output improves with guideline-directed medical therapy.[41][42] The recent DIGIT-heart failure trial demonstrated that digitoxin reduced the composite outcome of all-cause death or hospitalization for worsening heart failure in patients with advanced heart failure with reduced ejection fraction receiving guideline-directed therapy. Digitoxin has a more stable pharmacokinetic profile than digoxin because it undergoes hepatic elimination, exhibits higher protein binding, and distributes predominantly into skeletal muscle rather than relying on renal clearance. These findings highlight the importance of understanding differences in volume of distribution and elimination pathways among cardiac glycosides when selecting and dosing therapy in heart failure.[43] However, digitoxin is currently not approved by the FDA.
Specific Patient Populations
Hepatic impairment: Liver disease profoundly affects drug distribution through multiple interrelated mechanisms. Decreased hepatic albumin synthesis reduces PPB, increasing the free (unbound) fraction and the apparent Vd for highly protein-bound drugs (eg, phenytoin and warfarin). Portal-systemic shunting increases the oral bioavailability of drugs with a high hepatic extraction ratio by bypassing the first-pass metabolism. Ascites and peripheral edema expand the extracellular fluid (ECF) compartment, which may increase the Vd of hydrophilic drugs.
Impaired CYP450 metabolism (particularly CYP3A4, which is reduced in cirrhosis) can prolong drug half-life. The FDA guidance on pharmacokinetics in hepatic impairment recommends using the Child-Pugh classification to stratify dosing adjustments (Class A: mild [score 5–6], Class B: moderate [7–9], and Class C: severe [10–15]). The American Association for the Study of Liver Diseases' practice guidelines for the treatment of patients with hepatic disorders support the use of the Child-Pugh score for clinical assessment and prognostication, while acknowledging its limitations as a surrogate for hepatic metabolic capacity. Currently, no single biomarker reliably predicts hepatic drug clearance, and the Child-Pugh score remains the most widely accepted clinical tool.[23]
Vd-specific clinical implications in hepatic impairment include:
- For drugs with high PPB and low Vd (eg, warfarin: ~99% PPB, Vd≈0.14 L/kg), hypoalbuminemia increases the free drug fraction, leading to enhanced pharmacologic effect and bleeding risk even at normal total concentrations.
- For drugs with high hepatic extraction (eg, propofol and fentanyl), bioavailability may increase substantially due to portosystemic shunting; accordingly, the manufacturer of propofol recommends lower induction and maintenance doses in patients with cirrhosis.
- For drugs primarily metabolized by CYP3A4 (amiodarone, fentanyl, and amlodipine), dose reductions of 25% to 50% are typically required in Child-Pugh B or C patients.
- When Vd increases because of ascites or hypoalbuminemia, loading doses may need to be increased, whereas maintenance doses should generally be reduced because of impaired hepatic clearance.[44]
Renal impairment: The KDIGO established the definitive framework for drug dosing in kidney disease and is directly relevant to Vd.[9] Chronic kidney disease (CKD) and acute kidney injury (AKI) can alter Vd through several mechanisms, including fluid overload, which increases the Vd of hydrophilic drugs (eg, vancomycin and aminoglycosides); reduced PPB due to accumulation of uremic toxins, which increases the free fraction of drugs such as phenytoin and warfarin; and altered tissue binding.
Key KDIGO recommendations applicable to Vd-based dosing include:
- Loading doses generally do not require adjustment in stable CKD because Vd is not substantially altered.
- In AKI with fluid overload and/or sepsis, the Vd for hydrophilic antimicrobials may increase by approximately 25% to 50%, requiring higher initial doses.
- Maintenance doses must be adjusted according to the degree of reduction in creatinine clearance.
- Therapeutic drug monitoring (TDM) is essential for drugs with a narrow therapeutic index (eg, vancomycin, aminoglycosides, digoxin, and lithium)
- Drugs with small Vd and low PPB (gentamicin and lithium) are effectively removed by hemodialysis, whereas drugs with large Vd (amiodarone and digoxin) are poorly removed.
The 2020 American Society of Health-System Pharmacists (ASHP) and Infectious Diseases Society of America (IDSA) vancomycin guideline recommends using a Bayesian estimation of the area under the concentration-time curve to minimum inhibitory concentration ratio (AUC:MIC) to guide dosing, particularly in patients with CKD.[10][17][19][20]
Pregnancy considerations: Pregnancy induces significant pharmacokinetic changes that alter the Vd. Physiologic changes include plasma volume expansion of approximately 40% to 50% by the third trimester, increased total body water (up to about 8 L), decreased serum albumin due to dilutional hypoalbuminemia, increased cardiac output (30%-50%), and increased renal blood flow. These changes collectively increase Vd, particularly for hydrophilic drugs. The American College of Obstetricians and Gynecologists (ACOG) recognizes that pharmacokinetic changes during pregnancy may necessitate dose adjustments to maintain therapeutic efficacy and safety.[23][45][46][45][23]
Vd-specific implications in pregnancy include:
- Digoxin: Vd increases with plasma volume expansion, potentially lowering steady-state serum concentrations and may require dose increases with TDM guidance.
- Vancomycin and gentamicin: Vd may increase due to the expansion of the ECF. In addition, vancomycin clearance also increases due to augmented renal clearance, potentially requiring both higher loading and maintenance doses.
- Phenytoin: The free fraction increases due to reduced albumin; therefore, free phenytoin monitoring is strongly preferred over total levels.
- Warfarin: This is contraindicated during pregnancy due to teratogenic risk (historically classified as Category X) as per the DailyMed label and ACOG recommendations.
- Lithium: Vd increases with expanded plasma volume, and serum levels should be monitored more frequently, especially near term, when glomerular filtration rate begins to decline.
The FDA’s Pregnancy and Lactation Labeling Rule (effective from 2015) requires drug manufacturers to include dosing information during pregnancy when available.[6][16][23][46][23]
Breastfeeding considerations: Drug transfer into breast milk is influenced by multiple physicochemical properties, and Vd plays a specific role. The National Institutes of Health (NIH) LactMed database is a peer-reviewed, freely accessible resource for evidence-based information on drug levels in breast milk, infant serum concentrations, and potential adverse effects in nursing infants. A key pharmacokinetic parameter for breastfeeding risk assessment is the Relative Infant Dose (RID), which represents the weight-adjusted percentage of the maternal dose transferred to the infant through breast milk. In general, an RID below 10% is considered acceptable.[47]
Drugs with a high Vd tend to have a lower proportion of the total drug present in the plasma compartment available for transfer into milk, which may paradoxically reduce milk concentrations despite a high total body drug burden. However, Vd alone does not reliably predict drug transfer into milk. Other factors such as PPB, lipophilicity, molecular weight, and the milk-to-plasma (M/P) ratio must also be considered when evaluating drug safety during breastfeeding.[48][49]
Examples from drugs in this review using LactMed data include:
- Metoprolol (Vd 3.2–5.6 L/kg; ~224–392 L): This drug may concentrate in breast milk (M/P ~3), but RID is only 1% to 3% due to low absolute maternal plasma levels; therefore, it is usually considered compatible with breastfeeding.
- Fluoxetine (Vd 12–43 L/kg; ~840–3010 L): This drug has a large Vd; however, its active metabolite, norfluoxetine, has a long half-life (4–16 days) and may accumulate in breastfed infants. Reported adverse effects include colic, irritability, and poor feeding. As a result, sertraline or paroxetine may be preferred selective serotonin reuptake inhibitors (SSRIs) during lactation.
- Amiodarone (Vd 66 L/kg; ~4620 L): This drug should generally be avoided during breastfeeding because of its iodine content and extremely long half-life. Cases of thyroid toxicity have been reported in breastfed infants. If exposure occurs, monitoring of the infant’s cardiac and thyroid function may be considered.[50]
- Warfarin (Vd 0.14 L/kg; ~10 L; 99% protein binding): This drug has a low Vd; however, its very high PPB limits the amount of free drug available for transfer into breast milk. Consequently, warfarin is generally considered compatible with breastfeeding.[51]
Pediatric patients: Pediatric pharmacokinetics differs substantially from adult pharmacokinetics because of age-related changes in body composition, organ maturation, and PPB. Neonates have a higher proportion of total body water (approximately 70%-80% compared with about 60% in adults) and lower body fat, which can increase the Vd of hydrophilic drugs and decrease the Vd of lipophilic drugs. Serum concentrations of albumin and alpha-1 acid glycoprotein are also lower in neonates and infants, resulting in a higher free fraction of protein-bound drugs. In addition, hepatic CYP450 enzyme activity and renal glomerular filtration are immature at birth and mature at different rates throughout infancy and childhood. Therefore, pediatric-specific dosing considerations are required.[52][30]
Vd-specific implications in pediatric patients include:
- Aminoglycosides (eg, gentamicin and tobramycin) have a larger weight-normalized Vd in neonates (~0.4–0.5 L/kg versus 0.25 L/kg in adults) due to higher ECF volume, requiring higher per-kilogram doses (DailyMed: gentamicin label).
- Vancomycin Vd is also increased in neonates; dosing is typically adjusted based on both body weight and postnatal or gestational age according to institutional protocols.
- Phenytoin has a higher free fraction in neonates because of lower albumin concentrations and displacement by bilirubin; therefore, monitoring free phenytoin levels is particularly important.
- Digoxin Vd varies with age and is generally higher in infants and children than in adults; pediatric dosing is usually based on mcg/kg with TDM and PT/INR guidance.
- Theophylline has a higher Vd in neonates with slower clearance, contributing to the well-known increased risk of toxicity in this age group.
Allometric scaling approaches based on body weight and age are recommended for dose optimization.[53][30][54]
Older adults: Aging is associated with progressive pharmacokinetic changes that can alter the Vd. In older adults (aged ≥65), physiologic changes commonly include decreased total body water, increased body fat percentage, reduced lean muscle mass, lower serum albumin levels, and diminished hepatic and renal function. These changes may increase the Vd of lipophilic drugs (eg, amiodarone, propofol, and fluoxetine) while decreasing the Vd of hydrophilic drugs (eg, gentamicin and lithium). Reduced albumin levels can also increase the free fraction of highly protein-bound drugs, potentially enhancing pharmacologic effects and increasing the risk of toxicity at standard doses.
The American Geriatrics Society (AGS) 2023 Beers Criteria for potentially inappropriate medication (PIM) use in older adults identifies several drugs in this review as requiring special consideration in geriatric patients. For example, amiodarone is listed as a PIM due to the risk of thyroid dysfunction, pulmonary toxicity, QT prolongation, and peripheral neuropathy. Amiodarone's extremely large Vd and prolonged half-life in the geriatric population can exacerbate the duration of adverse effects. For digoxin, serum concentrations ≤1.0 ng/mL are recommended per the Beers Criteria, as older adults have reduced renal clearance and often decreased Vd (due to reduced lean mass), which predisposes them to toxicity. Phenytoin’s highly protein-bound status (90%) makes it particularly vulnerable to altered free fraction in geriatric patients with hypoalbuminemia.
Several Vd-informed dosing principles apply in geriatric patients:
- Lipophilic drugs with large Vd may accumulate more in increased adipose stores, prolonging half-life and duration of effect, thereby supporting the general approach of "start low, go slow."
- Hydrophilic drugs may have a reduced Vd due to decreased total body water, resulting in higher plasma concentrations at standard doses.
- TDM is particularly important in older patients for drugs with narrow therapeutic indices (eg, digoxin, phenytoin, theophylline, lithium, vancomycin, and aminoglycosides).
- The AGS Beers Criteria should also be incorporated into routine medication reviews for older adults to identify PIMs and guide deprescribing when risks outweigh benefits.[13]
Enhancing Healthcare Team Outcomes
Understanding that Vd is a proportionality constant that can yield different values depending on the drug concentration measured at a specific time in the body is important for clinical practice. This concept is relevant for the interprofessional healthcare team, including clinicians, intensivists, neonatologists, anesthesiologists, pharmacologists, and nurses, when prescribing, monitoring, or administering medications through any route of administration.[55]
Population pharmacokinetic models have been developed for several medications, including risperidone and paliperidone, providing detailed descriptions of their absorption, metabolism, and elimination. Future research should focus on the external validation of these models to facilitate their integration into clinical practice and support individualized dosing, ultimately improving treatment efficacy and safety across diverse patient populations.[56] Similarly, various other computational models have been developed to optimize individualized therapy with antimicrobials such as tigecycline and ampicillin-sulbactam.[57][58] Similarly, computational models have recently been developed across various therapeutic domains, including atogepant for migraine prophylaxis and tofacitinib for the treatment of active ankylosing spondylitis.[59][60]
The concept of Vd was first formalized in classical pharmacokinetic modeling and remains one of the most widely used parameters in clinical pharmacy practice. This is of paramount importance for calculating loading doses, estimating drug half-life, and interpreting plasma drug concentrations during TDM. The ASHP and the ACCP recognize the clinical utility of Vd in both acute care and ambulatory pharmacy practice settings.[4][61] American Society of Health-System Pharmacists. ASHP statement on the pharmacist’s role in clinical pharmacokinetic monitoring. Am J Health Syst Pharm. 1998;55:1726-1727. Available at: https://www.ashp.org. Accessed February 17, 2026.American College of Clinical Pharmacology. Clinical pharmacology practice guidelines and position papers. Available at: https://www.accp1.org. Accessed February 17, 2026.
Because medications have different volumes of distribution, appropriate dosing may depend on Vd to achieve adequate therapeutic drug concentrations, particularly when calculating loading doses and estimating the half-life (t1/2) of the drug. Understanding Vd helps bridge pharmacokinetic principles and clinical practice, making it valuable for the interprofessional healthcare team involved in patient care.
Media
(Click Image to Enlarge)
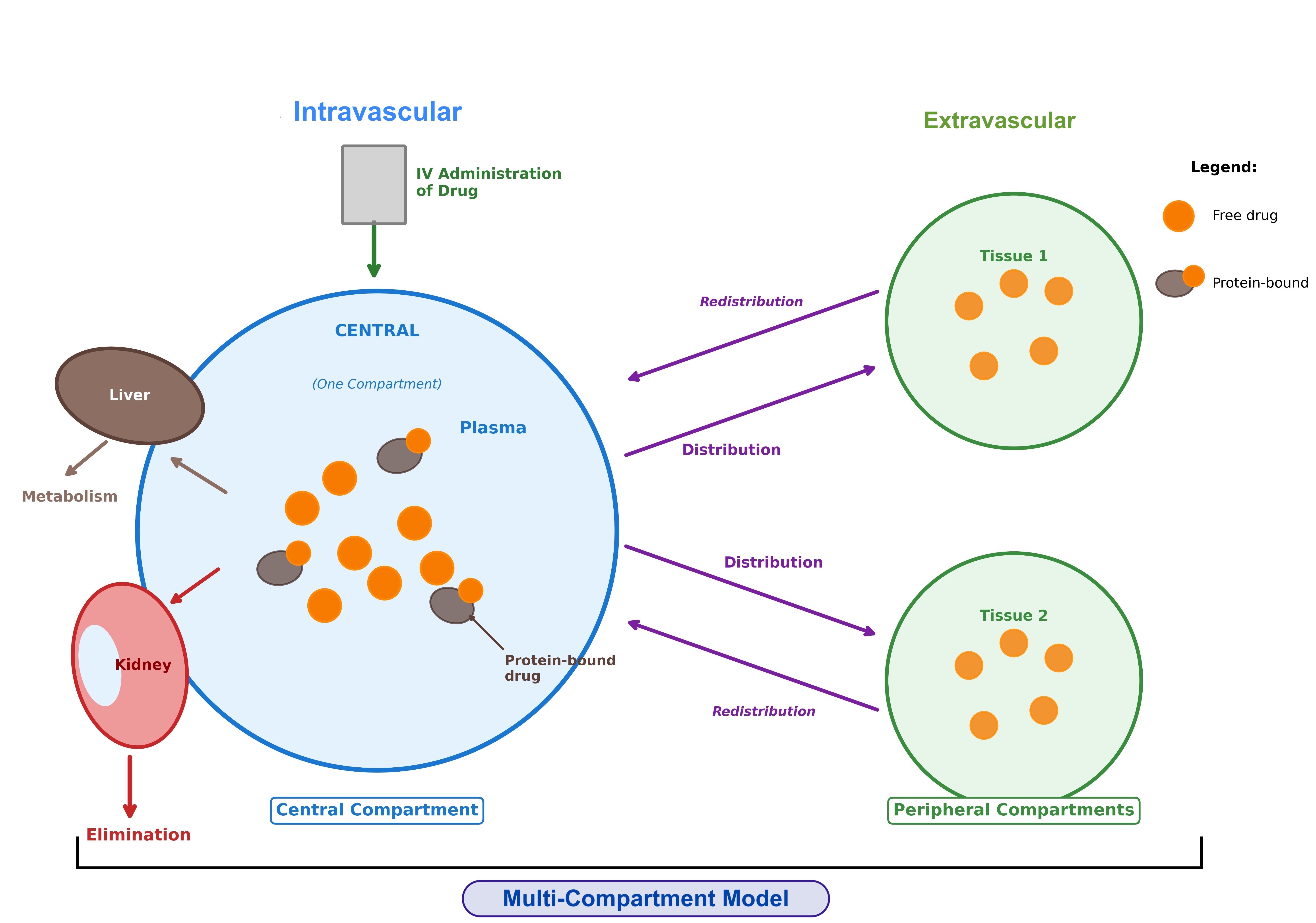
Single- Versus Multi-Compartment Model of Distribution.
Contributed by V Chauhan, MD
References
Oie S. Drug distribution and binding. Journal of clinical pharmacology. 1986 Nov-Dec:26(8):583-6 [PubMed PMID: 3793947]
Smith DA, Beaumont K, Maurer TS, Di L. Volume of Distribution in Drug Design. Journal of medicinal chemistry. 2015 Aug 13:58(15):5691-8. doi: 10.1021/acs.jmedchem.5b00201. Epub 2015 Apr 1 [PubMed PMID: 25799158]
Toutain PL, Bousquet-Mélou A. Volumes of distribution. Journal of veterinary pharmacology and therapeutics. 2004 Dec:27(6):441-53 [PubMed PMID: 15601439]
Level 3 (low-level) evidenceGreenblatt DJ. Volume of distribution - Again. Clinical pharmacology in drug development. 2014 Nov:3(6):419-20. doi: 10.1002/cpdd.173. Epub [PubMed PMID: 27129116]
Grover A, Benet LZ. Effects of drug transporters on volume of distribution. The AAPS journal. 2009 Jun:11(2):250-61. doi: 10.1208/s12248-009-9102-7. Epub 2009 Apr 28 [PubMed PMID: 19399628]
Holt K, Nagar S, Korzekwa K. Methods to Predict Volume of Distribution. Current pharmacology reports. 2019 Oct:5(5):391-399. doi: 10.1007/s40495-019-00186-5. Epub 2019 Jun 6 [PubMed PMID: 34168949]
Uges DR. Plasma or serum in therapeutic drug monitoring and clinical toxicology. Pharmaceutisch weekblad. Scientific edition. 1988 Oct 14:10(5):185-8 [PubMed PMID: 3060834]
Elkader A, Sproule B. Buprenorphine: clinical pharmacokinetics in the treatment of opioid dependence. Clinical pharmacokinetics. 2005:44(7):661-80 [PubMed PMID: 15966752]
Matzke GR, Aronoff GR, Atkinson AJ Jr, Bennett WM, Decker BS, Eckardt KU, Golper T, Grabe DW, Kasiske B, Keller F, Kielstein JT, Mehta R, Mueller BA, Pasko DA, Schaefer F, Sica DA, Inker LA, Umans JG, Murray P. Drug dosing consideration in patients with acute and chronic kidney disease-a clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney international. 2011 Dec:80(11):1122-37. doi: 10.1038/ki.2011.322. Epub 2011 Sep 14 [PubMed PMID: 21918498]
Rybak MJ, Le J, Lodise TP, Levine DP, Bradley JS, Liu C, Mueller BA, Pai MP, Wong-Beringer A, Rotschafer JC, Rodvold KA, Maples HD, Lomaestro BM. Therapeutic monitoring of vancomycin for serious methicillin-resistant Staphylococcus aureus infections: A revised consensus guideline and review by the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. American journal of health-system pharmacy : AJHP : official journal of the American Society of Health-System Pharmacists. 2020 May 19:77(11):835-864. doi: 10.1093/ajhp/zxaa036. Epub [PubMed PMID: 32191793]
Level 3 (low-level) evidenceBansal S, Nararyan R. Management of peritoneal dialysis in patients with obesity. Current opinion in nephrology and hypertension. 2026 Jan 1:35(1):101-107. doi: 10.1097/MNH.0000000000001124. Epub 2025 Oct 23 [PubMed PMID: 41133752]
Level 3 (low-level) evidenceCasati A, Putzu M. Anesthesia in the obese patient: pharmacokinetic considerations. Journal of clinical anesthesia. 2005 Mar:17(2):134-45 [PubMed PMID: 15809132]
By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. Journal of the American Geriatrics Society. 2023 Jul:71(7):2052-2081. doi: 10.1111/jgs.18372. Epub 2023 May 4 [PubMed PMID: 37139824]
Fan J, de Lannoy IA. Pharmacokinetics. Biochemical pharmacology. 2014 Jan 1:87(1):93-120. doi: 10.1016/j.bcp.2013.09.007. Epub 2013 Sep 17 [PubMed PMID: 24055064]
Level 3 (low-level) evidenceFaed EM. Protein binding of drugs in plasma, interstitial fluid and tissues: effect on pharmacokinetics. European journal of clinical pharmacology. 1981:21(1):77-81 [PubMed PMID: 7333350]
Andrade C. The Practical Importance of Half-Life in Psychopharmacology. The Journal of clinical psychiatry. 2022 Jul 25:83(4):. pii: 22f14584. doi: 10.4088/JCP.22f14584. Epub 2022 Jul 25 [PubMed PMID: 35900254]
Hollósy F, Valkó K, Hersey A, Nunhuck S, Kéri G, Bevan C. Estimation of volume of distribution in humans from high throughput HPLC-based measurements of human serum albumin binding and immobilized artificial membrane partitioning. Journal of medicinal chemistry. 2006 Nov 30:49(24):6958-71 [PubMed PMID: 17125249]
Bird HE, Huhn AS, Dunn KE. Fentanyl Absorption, Distribution, Metabolism, and Excretion: Narrative Review and Clinical Significance Related to Illicitly Manufactured Fentanyl. Journal of addiction medicine. 2023 Sep-Oct 01:17(5):503-508. doi: 10.1097/ADM.0000000000001185. Epub 2023 May 17 [PubMed PMID: 37788600]
Level 3 (low-level) evidenceKamp J, Olofsen E, Henthorn TK, van Velzen M, Niesters M, Dahan A, Ketamine Pharmacokinetic Study Group. Ketamine Pharmacokinetics. Anesthesiology. 2020 Dec 1:133(6):1192-1213. doi: 10.1097/ALN.0000000000003577. Epub [PubMed PMID: 32997732]
Mouton JWA, De Clercq A, De Paepe P, Petrovic M, Desmet T, Brüggemann RJ, Schouten JA, Jager NGL, De Cock PA. Pharmacokinetics and Target Attainment of Teicoplanin: A Systematic Review. Clinical pharmacokinetics. 2025 Apr:64(4):467-509. doi: 10.1007/s40262-025-01483-7. Epub 2025 Mar 10 [PubMed PMID: 40064832]
Level 1 (high-level) evidenceYim DS, Choi S. Predicting human pharmacokinetics from preclinical data: volume of distribution. Translational and clinical pharmacology. 2020 Dec:28(4):169-174. doi: 10.12793/tcp.2020.28.e19. Epub 2020 Dec 15 [PubMed PMID: 33425799]
Holford NH. Clinical pharmacokinetics and pharmacodynamics of warfarin. Understanding the dose-effect relationship. Clinical pharmacokinetics. 1986 Nov-Dec:11(6):483-504 [PubMed PMID: 3542339]
Level 3 (low-level) evidenceEl-Khateeb E, Darwich AS, Achour B, Athwal V, Rostami-Hodjegan A. Review article: time to revisit Child-Pugh score as the basis for predicting drug clearance in hepatic impairment. Alimentary pharmacology & therapeutics. 2021 Aug:54(4):388-401. doi: 10.1111/apt.16489. Epub 2021 Jul 4 [PubMed PMID: 34218453]
Iisalo E. Clinical pharmacokinetics of digoxin. Clinical pharmacokinetics. 1977 Jan-Feb:2(1):1-16 [PubMed PMID: 322907]
Hamilton D Sr, Nandkeolyar S, Lan H, Desai P, Evans J, Hauschild C, Choksi D, Abudayyeh I, Contractor T, Hilliard A. Amiodarone: A Comprehensive Guide for Clinicians. American journal of cardiovascular drugs : drugs, devices, and other interventions. 2020 Dec:20(6):549-558. doi: 10.1007/s40256-020-00401-5. Epub [PubMed PMID: 32166725]
Latini R, Tognoni G, Kates RE. Clinical pharmacokinetics of amiodarone. Clinical pharmacokinetics. 1984 Mar-Apr:9(2):136-56 [PubMed PMID: 6370540]
Freedman MD, Somberg JC. Pharmacology and pharmacokinetics of amiodarone. Journal of clinical pharmacology. 1991 Nov:31(11):1061-9 [PubMed PMID: 1753010]
Roberts FL, Dixon J, Lewis GT, Tackley RM, Prys-Roberts C. Induction and maintenance of propofol anaesthesia. A manual infusion scheme. Anaesthesia. 1988 Mar:43 Suppl():14-7 [PubMed PMID: 3259089]
Morse JD, Cortinez LI, Anderson BJ. Pharmacokinetic concepts for dexmedetomidine target-controlled infusion pumps in children. Paediatric anaesthesia. 2021 Sep:31(9):924-931. doi: 10.1111/pan.14235. Epub 2021 Jun 24 [PubMed PMID: 34085357]
Mahmood I. Dosing in children: a critical review of the pharmacokinetic allometric scaling and modelling approaches in paediatric drug development and clinical settings. Clinical pharmacokinetics. 2014 Apr:53(4):327-46. doi: 10.1007/s40262-014-0134-5. Epub [PubMed PMID: 24515100]
Ferguson-Sells L, Velez de Mendizabal N, Li B, Small D. Population Pharmacokinetics of Tadalafil in Pediatric Patients with Pulmonary Arterial Hypertension: A Combined Adult/Pediatric Model. Clinical pharmacokinetics. 2022 Feb:61(2):249-262. doi: 10.1007/s40262-021-01052-8. Epub 2021 Aug 11 [PubMed PMID: 34379314]
Luo D, Westhoff CL, Edelman AB, Natavio M, Stanczyk FZ, Jusko WJ. Altered pharmacokinetics of combined oral contraceptives in obesity - multistudy assessment. Contraception. 2019 Apr:99(4):256-263. doi: 10.1016/j.contraception.2018.12.009. Epub 2019 Jan 23 [PubMed PMID: 30684471]
Li ZR, Wang CY, Zhu X, Jiao Z. Population Pharmacokinetics of Levetiracetam: A Systematic Review. Clinical pharmacokinetics. 2021 Mar:60(3):305-318. doi: 10.1007/s40262-020-00963-2. Epub 2021 Jan 15 [PubMed PMID: 33447943]
Level 1 (high-level) evidenceZuckerman M, Greller HA, Babu KM. A Review of the Toxicologic Implications of Obesity. Journal of medical toxicology : official journal of the American College of Medical Toxicology. 2015 Sep:11(3):342-54. doi: 10.1007/s13181-015-0488-6. Epub [PubMed PMID: 26108709]
Czock D, Keller F, Rasche FM, Häussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clinical pharmacokinetics. 2005:44(1):61-98 [PubMed PMID: 15634032]
Level 3 (low-level) evidenceZamri PJ, Lim SMS, Sime FB, Roberts JA, Abdul-Aziz MH. A Systematic Review of Pharmacokinetic Studies of Colistin and Polymyxin B in Adult Populations. Clinical pharmacokinetics. 2025 May:64(5):655-689. doi: 10.1007/s40262-025-01488-2. Epub 2025 Apr 17 [PubMed PMID: 40246790]
Level 1 (high-level) evidenceWijnen NE, Touw DJ, Klein K, Kaspers GJL, Mian P. Pharmacokinetics of teicoplanin in paediatric patients-A systematic review of current literature. British journal of clinical pharmacology. 2026 Feb:92(2):396-421. doi: 10.1002/bcp.70309. Epub 2025 Oct 27 [PubMed PMID: 41145240]
Level 1 (high-level) evidenceSutar R, Atlani MK, Chaudhary P. Antipsychotics and hemodialysis: A systematic review. Asian journal of psychiatry. 2021 Jan:55():102484. doi: 10.1016/j.ajp.2020.102484. Epub 2020 Dec 3 [PubMed PMID: 33341539]
Level 1 (high-level) evidenceShammas FV, Dickstein K. Clinical pharmacokinetics in heart failure. An updated review. Clinical pharmacokinetics. 1988 Aug:15(2):94-113 [PubMed PMID: 3064953]
Ahmed A, Rich MW, Love TE, Lloyd-Jones DM, Aban IB, Colucci WS, Adams KF, Gheorghiade M. Digoxin and reduction in mortality and hospitalization in heart failure: a comprehensive post hoc analysis of the DIG trial. European heart journal. 2006 Jan:27(2):178-86 [PubMed PMID: 16339157]
CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The New England journal of medicine. 1987 Jun 4:316(23):1429-35 [PubMed PMID: 2883575]
Level 1 (high-level) evidencePitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. The New England journal of medicine. 1999 Sep 2:341(10):709-17 [PubMed PMID: 10471456]
Level 1 (high-level) evidenceBavendiek U, Großhennig A, Schwab J, Berliner D, Rieth A, Maier LS, Gaspar T, Thomas NH, Liu X, Schallhorn S, Angelini E, Soltani S, Rathje F, Sandu MA, Geller W, Hambrecht R, Zdravkovic M, Philipp S, Kosevic D, Nickenig G, Scheiber D, Winkler S, Becher PM, Lurz P, Hülsmann M, Wiesner S, Schröder C, Neuhaus B, Seltmann A, von der Leyen H, Veltmann C, Störk S, Böhm M, Koch A, Bauersachs J, DIGIT-HF Study Group. Digitoxin in Patients with Heart Failure and Reduced Ejection Fraction. The New England journal of medicine. 2025 Sep 25:393(12):1155-1165. doi: 10.1056/NEJMoa2415471. Epub 2025 Aug 29 [PubMed PMID: 40879434]
Idasiak-Piechocka I, Lewandowski D, Świgut W, Kalinowski J, Mikosza K, Suchowiejski P, Szałek E, Karbownik A, Miedziaszczyk M. Effect of hypoalbuminemia on drug pharmacokinetics. Frontiers in pharmacology. 2025:16():1546465. doi: 10.3389/fphar.2025.1546465. Epub 2025 Feb 20 [PubMed PMID: 40051558]
Feghali M, Venkataramanan R, Caritis S. Pharmacokinetics of drugs in pregnancy. Seminars in perinatology. 2015 Nov:39(7):512-9. doi: 10.1053/j.semperi.2015.08.003. Epub [PubMed PMID: 26452316]
Hansen H, Schäfer I, Schön G, Riedel-Heller S, Gensichen J, Weyerer S, Petersen JJ, König HH, Bickel H, Fuchs A, Höfels S, Wiese B, Wegscheider K, van den Bussche H, Scherer M. Agreement between self-reported and general practitioner-reported chronic conditions among multimorbid patients in primary care - results of the MultiCare Cohort Study. BMC family practice. 2014 Mar 1:15():39. doi: 10.1186/1471-2296-15-39. Epub 2014 Mar 1 [PubMed PMID: 24580758]
Spigset O. Using Relative Infant Dose to Assess Drug Exposure in Breastfed Infants. Basic & clinical pharmacology & toxicology. 2025 Oct:137(4):e70116. doi: 10.1111/bcpt.70116. Epub [PubMed PMID: 40976777]
Atkinson HC, Begg EJ, Darlow BA. Drugs in human milk. Clinical pharmacokinetic considerations. Clinical pharmacokinetics. 1988 Apr:14(4):217-40 [PubMed PMID: 3292101]
Abduljalil K, Faisal M. Impact of Milk pH and Fat Content on the Prediction of Milk-to-Plasma Ratio: Knowledge Gap and Considerations for Lactation Study Design and Interpretation. Clinical pharmacokinetics. 2024 Nov:63(11):1561-1572. doi: 10.1007/s40262-024-01432-w. Epub 2024 Oct 25 [PubMed PMID: 39453598]
. Amiodarone. Drugs and Lactation Database (LactMed®). 2006:(): [PubMed PMID: 29999620]
. Warfarin. Drugs and Lactation Database (LactMed®). 2006:(): [PubMed PMID: 30000196]
Wagner J, Abdel-Rahman SM. Pediatric pharmacokinetics. Pediatrics in review. 2013 Jun:34(6):258-69. doi: 10.1542/pir.34-6-258. Epub [PubMed PMID: 23729775]
Matson KL, Horton ER, Capino AC. Medication Dosing for Children With Overweight and Obesity. The journal of pediatric pharmacology and therapeutics : JPPT : the official journal of PPAG. 2024 Oct:29(5):550-553. doi: 10.5863/1551-6776-29.5.550. Epub 2024 Oct 14 [PubMed PMID: 39411409]
Santos BB, Heineck I, Negretto GW. USE OF WARFARIN IN PEDIATRICS: CLINICAL AND PHARMACOLOGICAL CHARACTERISTICS. Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao Paulo. 2017 Oct-Dec:35(4):375-382. doi: 10.1590/1984-0462/;2017;35;4;00008. Epub 2017 Sep 21 [PubMed PMID: 28977131]
Cropp CD, Beall J, Buckner E, Wallis F, Barron A. Interprofessional Pharmacokinetics Simulation: Pharmacy and Nursing Students' Perceptions. Pharmacy (Basel, Switzerland). 2018 Jul 20:6(3):. doi: 10.3390/pharmacy6030070. Epub 2018 Jul 20 [PubMed PMID: 30036982]
Carrascosa-Arteaga A, Nalda-Molina R, Más-Serrano P, Ramon-Lopez A. Population Pharmacokinetics of Risperidone and Paliperidone in Schizophrenia: A Systematic Review. Pharmaceuticals (Basel, Switzerland). 2025 May 8:18(5):. doi: 10.3390/ph18050698. Epub 2025 May 8 [PubMed PMID: 40430517]
Level 1 (high-level) evidenceDai A, Zheng F, Liu J, Chen Q, Zhou Z, Yu L, Yu Z, Guan Y. Population Pharmacokinetics of Tigecycline and Implications for Individualized Therapy Optimization: A Systematic Review. Drug design, development and therapy. 2025:19():10045-10060. doi: 10.2147/DDDT.S553622. Epub 2025 Nov 11 [PubMed PMID: 41244714]
Level 1 (high-level) evidencePoornachandran M, Harun SN, Yin TS, Yahaya N, Sheikh Ghadzi SM. Population Pharmacokinetic Models of Ampicillin-Sulbactam Among Adult Patients with Infectious Diseases: A Systematic Review. Therapeutic drug monitoring. 2026 Feb 1:48(1):60-70. doi: 10.1097/FTD.0000000000001380. Epub 2025 Sep 3 [PubMed PMID: 40903201]
Level 1 (high-level) evidenceSchlachter L, Stodtmann S, Voelkner A, Jonsson F, Lagraauw HM, Boinpally RR. Population Pharmacokinetics of Atogepant for the Prevention of Migraine. Clinical pharmacokinetics. 2026 Jan:65(1):149-164. doi: 10.1007/s40262-025-01566-5. Epub 2025 Nov 12 [PubMed PMID: 41222899]
Tsuchiwata S, Suzuki A, Wang Q, Kanik K, Fallon L, Menon S. Population pharmacokinetics of tofacitinib in patients with active ankylosing spondylitis. International journal of clinical pharmacology and therapeutics. 2026 Feb:64(2):57-65. doi: 10.5414/CP204781. Epub [PubMed PMID: 41355395]
Yu Z, Liu J, Yu H, Zhou L, Zhu J, Liang G, Yang Y, Zheng Y, Han Y, Xu J, Han G, Yu L, Zhao Y. Population pharmacokinetics and individualized dosing of vancomycin for critically ill patients receiving continuous renal replacement therapy: the role of residual diuresis. Frontiers in pharmacology. 2023:14():1298397. doi: 10.3389/fphar.2023.1298397. Epub 2023 Dec 29 [PubMed PMID: 38223197]