Introduction
Lymphocytic hypophysitis is a rare inflammatory disorder of the pituitary gland that presents with features of hypopituitarism secondary to a sellar mass lesion.[1] The condition is characterized by infiltration of the pituitary gland with T and B lymphocytes.[2] This autoimmune condition is the most frequent histopathologic subtype of primary hypophysitis.[3] Endocrine symptoms are due to pituitary dysfunction and may include central diabetes insipidus, anterior pituitary hormone deficiencies, hyperprolactinemia, or hypoprolactinemia.[4]
Patients may also present with symptoms of mass effect, such as headaches or visual field disturbances, which can mimic a pituitary adenoma. Early recognition is crucial for guiding appropriate hormonal replacement and preventing unnecessary surgical intervention.[5] Because lymphocytic hypophysitis is relatively rare, diagnosis and treatment can be challenging.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Hypophysitis is categorized based on etiology as primary or secondary.[6] Primary hypophysitis is further categorized based on histology as lymphocytic, granulomatous, xanthomatous, immunoglobulin (Ig) G4-related, or mixed, and based on anatomy as lymphocytic adenohypophysitis, lymphocytic infundibuloneurohypophysitis, or lymphocytic panhypophysitis.[4][7] Lymphocytic adenohypophysitis affects the anterior pituitary gland, lymphocytic infundibuloneurohypophysitis affects the posterior pituitary gland, and lymphocytic panhypophysitis affects both the anterior and posterior lobes of the pituitary gland.[2] The condition is characterized by lymphocytic infiltration of the pituitary gland, most commonly involving CD4+ and CD8+ T cells. Antipituitary antibodies are also involved, although these are not specific or sensitive enough for routine clinical use. A combination of genetic predisposition, environmental factors, and pituitary-specific antigens, such as α-enolase, may trigger the autoimmune process.[8]
Although primary hypophysitis describes inflammation of the pituitary gland, secondary hypophysitis refers to pituitary inflammation caused by diseases such as sarcoidosis, hemochromatosis, amyloidosis, granulomatosis with polyangiitis, tuberculosis, syphilis, or medications. Immunomodulatory drugs, including immune checkpoint inhibitors such as cytotoxic T-lymphocyte–associated protein 4 and programmed death 1 inhibitors, are recognized causes.[6] Lymphocytic hypophysitis was first described in 1962 by Goudie and Pinkerton, who noted it as the most common form of primary hypophysitis and associated it with autoimmune pathogenesis.[2] As a result of this discovery, the condition is sometimes called autoimmune hypophysitis.[3]
Epidemiology
The prevalence of all types of hypophysitis is low, with an incidence of approximately 1 in 9 million.[2] However, this figure may underestimate the true disease incidence, owing to increased recognition of IgG4-related disease and the recent use of immune checkpoint inhibitors for cancer treatments, which are associated with endocrine effects on the pituitary gland.[4] Lymphocytic hypophysitis accounts for the majority of primary hypophysitis cases and occurs more commonly in women, particularly during late pregnancy and the postpartum period.
Although initially believed to affect only peripartum women, lymphocytic hypophysitis is now recognized to occur in nonpregnant women and men. Female-to-male ratio ranges from 3:1 to 8:1, with a median age at diagnosis in the third to fourth decade of life.[9] Although cases have been reported in children and older adults, the mean age of diagnosis is 44.7 years in men and 34 years in women.[5] Hypophysitis is associated with other autoimmune diseases in 20% to 50% of the reported cases.[6]
Histopathology
Although histology provides a definitive diagnosis of lymphocytic hypophysitis, it requires a biopsy or resection of sellar tissue, which is high-risk and not routinely performed.[4] The histologic evaluation demonstrates infiltration of the adenohypophysis with lymphocytes, plasma cells, and macrophages. T and B lymphocytes infiltrating the pituitary gland can also form lymphoid follicles with a germinal center.[2] Histological stains can be used to identify inflammatory cells and assess the degree of lymphocytic infiltration.[4]
The degree of lymphocytic infiltration corresponds to the degree of pituitary gland enlargement, which exerts pressure on nearby structures, causing headaches, visual problems, and subclinical hypopituitarism. Subclinical hypopituitarism may resolve if the pituitary gland tissue is not destroyed. Fibrosis of the pituitary gland can also develop as the disease progresses.[9] If the pituitary tissue is destroyed by extensive lymphocytic infiltration, it is replaced by fibrous tissue, and symptoms of partial or complete hypopituitarism may develop (see Image. Lymphocytic Hypophysitis).
The typical histological pattern of the pituitary gland is composed of cords and clusters of polygonal endocrine cells supported by a delicate reticulin network and a rich sinusoidal capillary plexus. The primary cell types include acidophils (somatotrophs and lactotrophs), basophils (corticotrophs, thyrotrophs, and gonadotrophs), and chromophobes (less intensely staining cells). These cells are distributed in a characteristic pattern, with acidophils more prominent in the lateral wings, basophils in the central wedge, and chromophobes scattered throughout. The pars distalis is separated from the neurohypophysis by the pars intermedia, which is rudimentary in humans and contains colloid-filled cysts lined by melanotrophs and folliculostellate cells. The pars tuberalis, which wraps around the pituitary stalk, mainly comprises gonadotrophs but also includes other hormone-producing cells. Please see Statpearls' companion resource, "Anatomy, Head and Neck, Pituitary Gland," for further information (see Image. Normal Pituitary Gland Cells, Histology).
History and Physical
Although lymphocytic hypophysitis was initially believed to affect only the anterior pituitary, it can also involve the posterior pituitary and pituitary stalk, causing inflammation and lymphocyte, macrophage, and plasma cell infiltration.[9] Lymphocytic hypophysitis can lead to deficiencies in 1 or more pituitary hormones, causing central diabetes insipidus if the posterior pituitary gland is affected, and secondary adrenal insufficiency and secondary hypothyroidism if the anterior pituitary gland is affected.[6][9] Other conditions that can occur due to pituitary inflammation include hypogonadotropic hypogonadism and growth hormone deficiency.[2] Enlargement of the pituitary gland can compress the optic chiasm, leading to neuro-ophthalmic manifestations such as altered vision and color perception.[4][10]
Hypophysitis typically presents with symptoms of pituitary hormone deficiencies and may be accompanied by headaches or visual disturbances due to mass effect. Other common symptoms include nausea, vomiting, fatigue, dizziness, loss of libido, and amenorrhea. Approximately 50% of patients report headaches, and 10% to 30% experience visual symptoms. Hormonal deficits vary based on the extent and cause of pituitary damage. The co-occurrence of both anterior pituitary dysfunction and diabetes insipidus strongly suggests an inflammatory or infiltrative process affecting the pituitary stalk rather than a pituitary adenoma. In lymphocytic and immunotherapy-induced hypophysitis, corticotrophs are initially affected, followed by gonadotrophs and thyrotrophs, a pattern opposite to that observed in pituitary adenomas. Eventually, pituitary destruction can occur due to infiltration and inflammation, and hypopituitarism can affect almost all pituitary hormones.[2][8][9][11]
Evaluation
Lymphocytic hypophysitis is a diagnosis of exclusion, and histopathology with tissue biopsy is required for definitive diagnosis.[3] However, clinical, laboratory data, and imaging can contribute to the diagnosis.[10] Patients often present with symptoms of hypopituitarism and should undergo pituitary hormone function evaluation.[9] Relevant laboratory studies include prolactin, adrenocorticotropic hormone, cortisol, insulin-like growth factor 1, growth hormone, thyroid-stimulating hormone, free thyroxine, testosterone or estradiol, luteinizing hormone, and follicle-stimulating hormone. Diabetes insipidus can be diagnosed by obtaining electrolyte levels, measuring antidiuretic hormone levels, assessing serum and urine osmolality, and performing water deprivation testing if necessary.
Approximately 20% to 50% of individuals with lymphocytic hypophysitis have other autoimmune diseases.[6] Antipituitary and antihypothalamus antibodies have also been identified, but testing for these antibodies may not be practical.[2] The initial laboratory evaluation for suspected hypophysitis includes a complete blood count, erythrocyte sedimentation rate, C-reactive protein, calcium, creatinine, urinalysis, and alanine aminotransferase. Clinical findings guide additional testing and may include IgG4 levels, angiotensin-converting enzyme levels, antineutrophil cytoplasmic antibody titers, antinuclear antibody titers, lactate dehydrogenase levels, β2-microglobulin levels, α-fetoprotein levels, human chorionic gonadotropin levels, and tuberculosis screening. Although systemic disease features may prompt these tests, their diagnostic utility is limited. Pit-1, a pituitary-specific transcription factor, has recently been identified as a potential autoimmune target in lymphocytic hypophysitis, but testing for Pit-1 antibodies is not yet widely accessible.[12][13] Cerebrospinal fluid analysis may be useful when initial tests are inconclusive and there is concern for infection or malignancy. Cerebrospinal fluid tests include cytology, flow cytometry, angiotensin-converting enzyme levels, human chorionic gonadotropin levels, α-fetoprotein levels, and bacterial cultures.[14]
Gadolinium-enhanced magnetic resonance imaging (MRI) of the pituitary is useful for distinguishing lymphocytic hypophysitis from pituitary adenoma. Pituitary adenoma is typically characterized by asymmetrical pituitary enlargement with a deviated stalk, whereas lymphocytic hypophysitis demonstrates a symmetrical pituitary gland and pituitary stalk enlargement without stalk deviation. MRI findings may demonstrate a homogeneously intense pituitary with dural enhancement (dural tail) and arachnoid enhancement in lymphocytic hypophysitis.[4][9][15] In contrast, pituitary adenomas do not demonstrate enhancement of the dura or arachnoid. Despite these findings, radiologic imaging cannot always distinguish between an adenoma and lymphocytic hypophysitis; up to 40% of patients with lymphocytic hypophysitis are initially misdiagnosed as having a pituitary macroadenoma.[3]
Many of these patients undergo unnecessary surgery because symptoms are attributed to a pituitary adenoma.[9] Surgery should be avoided if a diagnosis can be made without the need for histopathology, and it is recommended if pituitary inflammation and infiltration are causing visual loss.[16] Biopsy criteria are not standardized in adults; however, the United Kingdom pediatric guidelines recommend biopsy when the diagnosis remains uncertain after comprehensive testing, particularly if the pituitary stalk is 6.5 mm or larger, or when clinical worsening occurs, such as in the case of progressive hormonal deficits, structural changes, or new visual symptoms.[14]
In response to frequent misdiagnoses, the Gutenberg scoring system was developed to accurately identify lymphocytic hypophysitis. This scoring system incorporates MRI findings to aid in the diagnosis of this disease preoperatively:
- Age <30 years: −1
- Current pregnancy or within 6 months postpartum: −4
- Increased stalk size: −5
- Increased gadolinium enhancement: −1
- Loss of pituitary bright spot: −2
- Pituitary volume >6 cm3: +2
- Sphenoid mucosal thickening: +2
- Heterogeneous gadolinium enhancement: +1
- Asymmetrical sellar enlargement: +3 [3]
A total score greater than 1 is suggestive of a pituitary adenoma. In contrast, a score of 0 or less is suggestive of lymphocytic hypophysitis, with a specificity of 99%, sensitivity of 92%, and positive and negative predictive values of 97%.[3]
Treatment / Management
The primary goal in treating lymphocytic hypophysitis is to manage pituitary hormone deficiencies and any mass effects due to pituitary gland enlargement. Conservative treatment can include regular surveillance without intervention or anti-inflammatory therapy. Surgery or radiotherapy is rarely required. However, pituitary function needs to be assessed regularly, and hormone deficiencies should be treated accordingly.[17] Glucocorticoids are commonly used to treat primary hypophysitis due to their anti-inflammatory effects. However, spontaneous improvement in pituitary inflammation may also occur without treatment. Results from a German study showed that nearly half of patients observed without therapy had improved imaging findings, and about one-third experienced partial hormonal recovery, primarily in vasopressin and adrenocorticotropic hormone function.[18] Patients should also be assessed and treated for diabetes insipidus. Although spontaneous resolution can occur, corticosteroid therapy is the primary treatment for most patients.[4] If observation is the chosen management approach, regular reassessment of pituitary function is essential to monitor for recovery.
If vision is affected, mass effect involves nearby vital structures, or histological confirmation is required, surgery becomes necessary. Patients sometimes require long-term hormone replacement therapy after neurosurgery, depending on the extent of surgery and patient factors. However, corticosteroid therapy, which has an anti-inflammatory effect and can decrease the size of the mass lesion, could potentially restore pituitary function and reduce the need for lifelong hormone therapy if recurrence after treatment does not occur. However, there is no consensus regarding the optimal dose and duration of glucocorticoid therapy. A study in Germany treated 32 patients with prednisolone-equivalent glucocorticoids at daily doses ranging from 20 to 500 mg for durations of 4 days to 1 year.[18]
Corticosteroids should be tapered based on the response and adverse effects, including weight gain, Cushing syndrome, edema, glaucoma, psychiatric symptoms, and diabetes mellitus. Approximately 38% of patients experience a recurrence despite an initial response to corticosteroids. For patients who do not improve with corticosteroids or experience recurrent disease after treatment, immunosuppressive medications such as methotrexate, azathioprine, cyclosporine, rituximab, and mycophenolate mofetil may be considered.[16][19][20] Dopamine agonists such as cabergoline and bromocriptine have also been effective in patients with hyperprolactinemia due to pituitary inflammation.[4] For patients experiencing severe headache and notable mass effect, high-dose glucocorticoids are the preferred initial treatment. Although lymphocytic hypophysitis typically responds to corticosteroid therapy, the response is generally less robust in IgG4-related disease.[21](B3)
Surgery is reserved for patients with visual symptoms, ophthalmoplegia, mass effect, or equivocal imaging findings requiring histology for diagnosis. Surgery provides histologic confirmation for diagnosis and excludes a pituitary tumor. The surgical approach is typically transsphenoidal or transcranial partial resection of the pituitary lesion or decompression of the mass. Recurrence occurs in 11% to 25% of cases with no relationship to the extent of the resection. Surgical complications include postoperative meningitis and rhinorrhea. Occasionally, glucocorticoid therapy and surgery are both used.[18] If surgery, corticosteroids, and immunosuppressive treatment do not improve symptoms, fractionated radiotherapy may be considered a last resort.[22](A1)
Differential Diagnosis
Obtaining a detailed history and performing a thorough physical examination are essential for diagnosing lymphocytic hypophysitis. However, without a tissue biopsy, evidence of pituitary mass, or symptoms of hypopituitarism, the differential diagnosis is broad.[10] The differential diagnosis of hypophysitis encompasses inflammatory, autoimmune, infectious, neoplastic, and physiological conditions, including pituitary adenoma, craniopharyngioma, Rathke cleft cyst, germinoma, pituitary apoplexy, and pituitary metastasis.[3][23][24] Drug-induced hypophysitis, particularly from immune checkpoint inhibitor medications and other immunotherapies, is diagnosed based on recent treatment history.
Autoimmune disorders, such as thyroid disease, type 1 diabetes, polyglandular syndromes, and connective tissue diseases, including systemic lupus erythematosus and Sjögren disease, may present with systemic symptoms and require disease-specific serologic testing. Inflammatory bowel disease, sarcoidosis, and infiltrative diseases such as Langerhans cell histiocytosis and Erdheim-Chester disease may also mimic hypophysitis, often with multisystem involvement and characteristic imaging or biopsy findings. IgG4-related disease typically affects older men and may involve other organs; it is generally diagnosed through elevated serum IgG4 levels and tissue biopsy. Vasculitides, such as temporal arteritis or granulomatosis with polyangiitis, can affect the pituitary and present with systemic inflammation and vascular symptoms.
Infections such as tuberculosis or fungal disease are rare but possible, especially in immunocompromised patients. Paraneoplastic syndromes and sellar or parasellar tumors (eg, craniopharyngioma, germinoma, and meningioma) must be excluded because they can mimic or cause secondary hypophysitis. Other considerations include pituitary metastases, apoplexy, lymphoproliferative malignancies, Sheehan syndrome, and physiological pituitary hypertrophy, each with distinguishing clinical and radiologic features.[4][6][7][20] Accurate diagnosis relies on clinical context, laboratory testing, imaging, and occasionally, biopsy.
Prognosis
Lymphocytic hypophysitis often presents with symptoms and clinical features similar to those of a pituitary adenoma.[9] Previously, diagnoses were made through autopsy; however, due to advancements in imaging, the number of reported cases has increased over the past 3 decades. Because of lymphocytic infiltration of the pituitary gland and subsequent inflammation, hypopituitarism involving multiple pituitary hormones can occur. Treatment varies based on symptoms and may include expectant observation to monitor for spontaneous remission, glucocorticoid therapy, immunosuppressive agents, partial resection of the pituitary lesion, or surgical decompression if nearby vital structures are compromised by mass effect.[9] Diagnosis without histopathological confirmation can be difficult because some patients may be asymptomatic; however, most present with headaches, visual defects, or other endocrine-related symptoms.[24]
Diagnosis frequently lags behind symptom onset.[25] Recurrence can occur even after medical and surgical therapy, requiring lifelong hormone replacement therapy.[16] Timely diagnosis and appropriate treatment of hormone deficiency, especially adrenal insufficiency, is essential due to the high rate of morbidity and mortality associated with this condition.[17] Despite the complications and diagnostic challenges, lymphocytic hypophysitis can be effectively managed with corticosteroids, surgery, immunosuppressive agents, and regular surveillance, resulting in a favorable prognosis.[25]
The prognosis for lymphocytic hypophysitis is generally favorable, particularly with respect to resolution of mass effect and overall survival. However, most patients develop permanent pituitary hormone deficiencies that require lifelong replacement therapy. Spontaneous regression of the inflammatory mass may occur, especially in milder cases, making observation a reasonable approach in asymptomatic patients without significant mass effect. The presence of anterior pituitary dysfunction or diabetes insipidus at diagnosis often predicts irreversible hypopituitarism. Continued surveillance is crucial to detect recurrence and adjust hormone therapy as needed.[20][26]
Complications
Lymphocytic hypophysitis requires prompt recognition, as significant complications can occur.[25] Many patients develop anterior pituitary hormone deficiencies and diabetes insipidus, resulting in panhypopituitarism. Prolactin levels may be decreased, normal, or increased. Cortisol and adrenocorticotropic hormone levels should be obtained to diagnose and treat adrenal insufficiency, which may result from lymphocytic hypophysitis.[17]
Because secondary adrenal insufficiency progresses rapidly, cosyntropin stimulation test results may appear normal early in the disease course, as adrenal gland atrophy has not yet occurred. An insulin-induced hypoglycemia test may be required to assess adrenal insufficiency at an earlier stage. Other hormone deficiencies can occur, and laboratory evaluation should include prolactin, insulin-like growth factor 1, growth hormone, thyroid-stimulating hormone, free thyroxine, testosterone or estradiol, luteinizing hormone, follicle-stimulating hormone, electrolyte levels, antidiuretic hormone, and serum and urine osmolality.[4] Pituitary gland enlargement can also cause neurological complications and visual field defects due to mass effect on the optic nerve, optic chiasm, and cavernous sinus (cranial nerves III, IV, and VI). Visual field defects can include impaired color vision perception, ophthalmoplegia, and diplopia.[4][9][10]
Deterrence and Patient Education
Lymphocytic hypophysitis is a rare autoimmune condition characterized by lymphocytic infiltration and inflammation of the pituitary gland.[2] The condition occurs more frequently in women than in men, with a mean age at diagnosis of 34 years in women and 45 years in men.[9] Common symptoms include headache, vision disturbance, nausea, vomiting, dizziness, fatigue, decreased libido, and amenorrhea. In most cases, laboratory evaluation and brain MRI are performed, and treatment is tailored to the individual patient.[4] Management may include supportive care, corticosteroids, immunosuppressive medications, and surgery, especially for vision loss.[16]
Patients diagnosed with lymphocytic hypophysitis should be informed that the inflammatory process affecting the pituitary gland may improve or resolve over time, but frequently leads to permanent hormone deficiencies requiring lifelong hormone replacement therapy. These hormonal imbalances can affect energy levels, metabolism, stress response, water balance, and reproductive function. Patients should be counseled on symptoms of hormone deficiency, such as fatigue, dizziness, excessive thirst, or changes in appetite, body weight, or menstruation. Regular follow-up with the healthcare team is essential to monitor hormone levels, adjust treatment, and ensure overall well-being. With proper management and education, patients can lead healthy and productive lives.[25]
Enhancing Healthcare Team Outcomes
Optimal care for lymphocytic hypophysitis requires a coordinated interprofessional team effort. Primary care clinicians, endocrinologists, neurosurgeons, radiologists, ophthalmologists, pharmacists, and nurses play critical roles. Clinicians must integrate clinical, radiologic, and laboratory findings to guide diagnosis and therapy. Radiologists contribute diagnostic precision, whereas endocrinologists oversee hormonal evaluation and replacement.
Nurses and pharmacists ensure medication adherence, monitor for adverse effects, and educate patients on long-term care needs. Effective interprofessional communication, shared decision-making, and ethical responsibility are crucial for enhancing patient-centered care, improving safety and outcomes, and ensuring timely treatment. This collaborative strategy fosters accurate diagnosis, ensures continuity of care, and improves the quality of life for patients with this rare condition.[5][24][20]
Media
(Click Image to Enlarge)
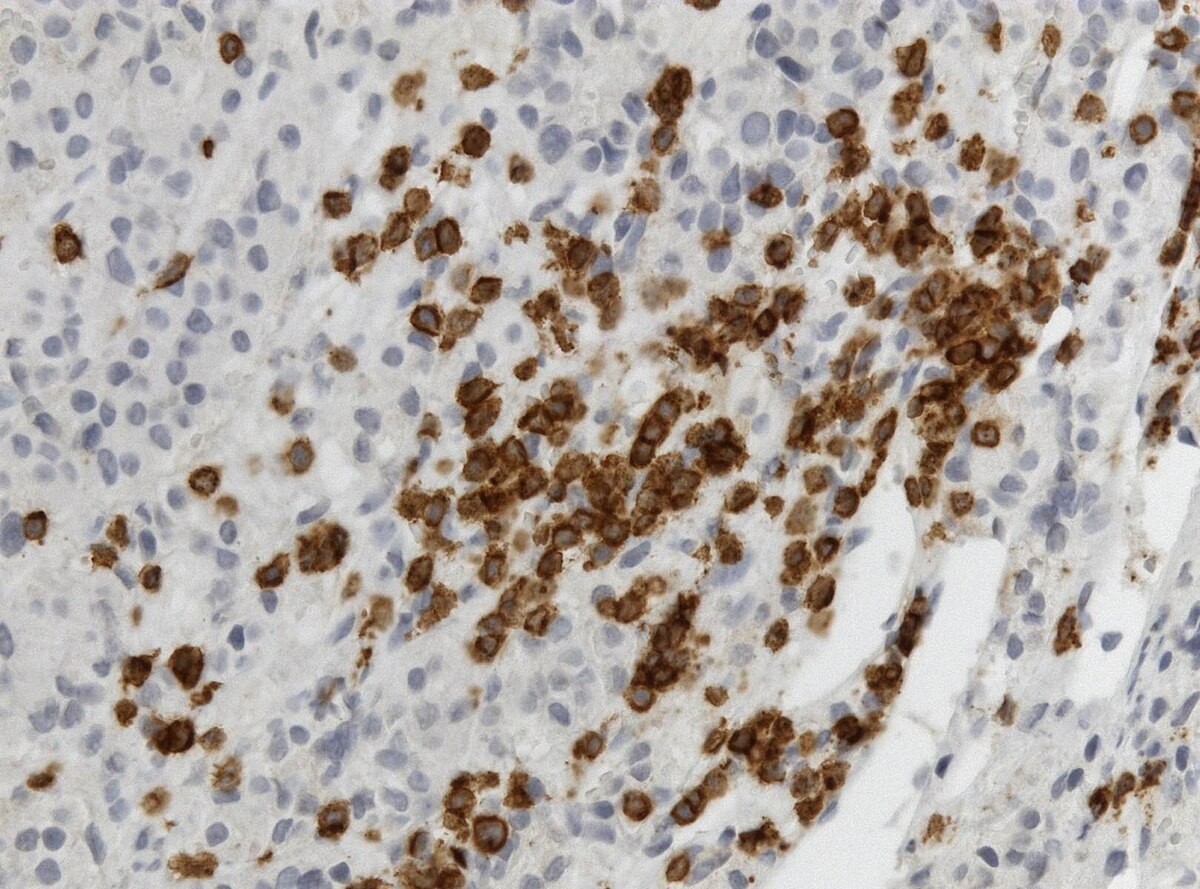
Lymphocytic Hypophysitis. CD3 immunohistochemistry showing T cells (brown) in lymphocytic hypophysitis.
Jensflorian, Public Domain, via Wikimedia Commons
{kind=link}
(Click Image to Enlarge)
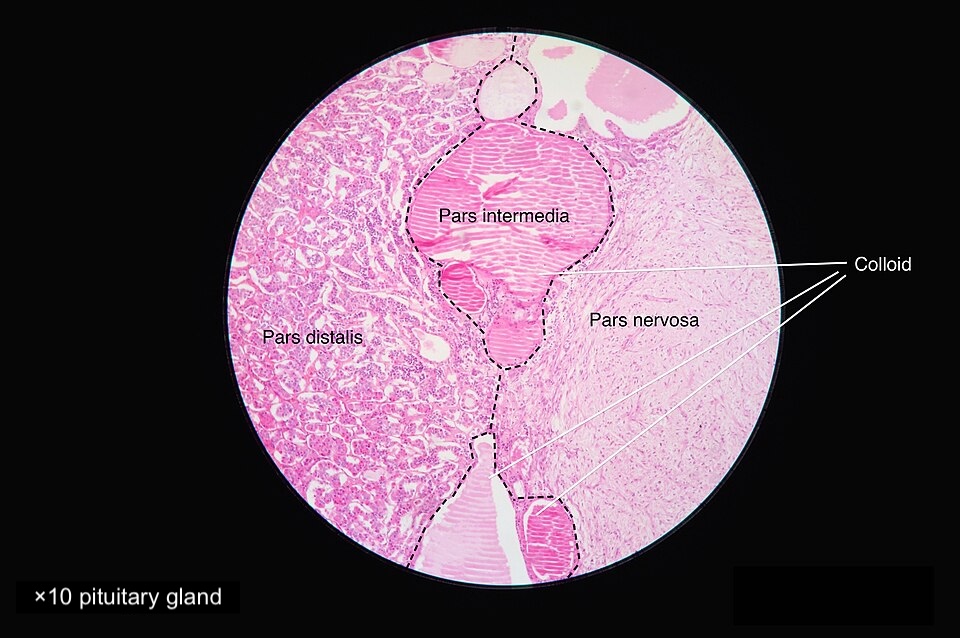
Normal Pituitary Gland Cells, Histology. This image demonstrates the typical histological appearance of pituitary gland cells.
Athikhun.suw, Public Domain, via Wikimedia Commons
{kind=link}
References
Molitch ME, Gillam MP. Lymphocytic hypophysitis. Hormone research. 2007:68 Suppl 5():145-50. doi: 10.1159/000110611. Epub 2007 Dec 10 [PubMed PMID: 18174733]
Honegger J, Schlaffer S, Menzel C, Droste M, Werner S, Elbelt U, Strasburger C, Störmann S, Küppers A, Streetz-van der Werf C, Deutschbein T, Stieg M, Rotermund R, Milian M, Petersenn S, Pituitary Working Group of the German Society of Endocrinology. Diagnosis of Primary Hypophysitis in Germany. The Journal of clinical endocrinology and metabolism. 2015 Oct:100(10):3841-9. doi: 10.1210/jc.2015-2152. Epub 2015 Aug 11 [PubMed PMID: 26262437]
Nanda A, Savardekar AR, Patra DP. Diagnosis and management of lymphocytic hypophysitis: A synopsis on current perspective. Neurology India. 2018 Mar-Apr:66(2):405-406. doi: 10.4103/0028-3886.227282. Epub [PubMed PMID: 29547162]
Level 3 (low-level) evidenceJoshi MN, Whitelaw BC, Carroll PV. MECHANISMS IN ENDOCRINOLOGY: Hypophysitis: diagnosis and treatment. European journal of endocrinology. 2018 Sep:179(3):R151-R163. doi: 10.1530/EJE-17-0009. Epub 2018 Jun 7 [PubMed PMID: 29880706]
Patil AA, Patil P, Walke V. Lymphocytic Hypophysitis: An Underrated Disease. Journal of mid-life health. 2022 Jul-Sep:13(3):254-256. doi: 10.4103/jmh.jmh_32_21. Epub 2023 Jan 14 [PubMed PMID: 36950203]
Chang LS, Barroso-Sousa R, Tolaney SM, Hodi FS, Kaiser UB, Min L. Endocrine Toxicity of Cancer Immunotherapy Targeting Immune Checkpoints. Endocrine reviews. 2019 Feb 1:40(1):17-65. doi: 10.1210/er.2018-00006. Epub [PubMed PMID: 30184160]
Can S, Tihan T, Alele J, Robbins RJ. Giant-cell granulomatous hypophysitis. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 1998 Jan-Feb:4(1):41-7 [PubMed PMID: 15251764]
Frasca F, Piticchio T, Le Moli R, Malaguarnera R, Campennì A, Cannavò S, Ruggeri RM. Recent insights into the pathogenesis of autoimmune hypophysitis. Expert review of clinical immunology. 2021 Nov:17(11):1175-1185. doi: 10.1080/1744666X.2021.1974297. Epub 2021 Sep 6 [PubMed PMID: 34464545]
Rumana M, Kirmani A, Khursheed N, Besina S, Khalil M. Lymphocytic hypophysitis with normal pituitary function mimicking a pituitary adenoma: a case report and review of literature. Clinical neuropathology. 2010 Jan-Feb:29(1):26-31 [PubMed PMID: 20040330]
Level 3 (low-level) evidenceYuen KCJ, Popovic V, Trainer PJ. New causes of hypophysitis. Best practice & research. Clinical endocrinology & metabolism. 2019 Apr:33(2):101276. doi: 10.1016/j.beem.2019.04.010. Epub 2019 Apr 26 [PubMed PMID: 31078416]
Oguz SH, Soylemezoglu F, Sendur SN, Mut M, Oguz KK, Dagdelen S, Erbas T. Clinical Characteristics, Management, and Treatment Outcomes of Primary Hypophysitis: A Monocentric Cohort. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2020 Apr:52(4):220-227. doi: 10.1055/a-1113-7777. Epub 2020 Apr 8 [PubMed PMID: 32268423]
Guaraldi F, Giordano R, Grottoli S, Ghizzoni L, Arvat E, Ghigo E. Pituitary Autoimmunity. Frontiers of hormone research. 2017:48():48-68. doi: 10.1159/000452905. Epub 2017 Feb 28 [PubMed PMID: 28245451]
Ricciuti A, De Remigis A, Landek-Salgado MA, De Vincentiis L, Guaraldi F, Lupi I, Iwama S, Wand GS, Salvatori R, Caturegli P. Detection of pituitary antibodies by immunofluorescence: approach and results in patients with pituitary diseases. The Journal of clinical endocrinology and metabolism. 2014 May:99(5):1758-66. doi: 10.1210/jc.2014-1049. Epub 2014 Feb 25 [PubMed PMID: 24606106]
Cerbone M, Visser J, Bulwer C, Ederies A, Vallabhaneni K, Ball S, Kamaly-Asl I, Grossman A, Gleeson H, Korbonits M, Nanduri V, Tziaferi V, Jacques T, Spoudeas HA. Management of children and young people with idiopathic pituitary stalk thickening, central diabetes insipidus, or both: a national clinical practice consensus guideline. The Lancet. Child & adolescent health. 2021 Sep:5(9):662-676. doi: 10.1016/S2352-4642(21)00088-2. Epub 2021 Jun 30 [PubMed PMID: 34214482]
Level 3 (low-level) evidenceJu JS, Cui T, Zhao J, Chen JL, Ju HB. Clinical presentation and magnetic resonance imaging characteristics of lymphocytic hypophysitis: a systematic review with meta-analysis. Archives of medical science : AMS. 2023:19(4):976-986. doi: 10.5114/aoms/144628. Epub 2021 Dec 14 [PubMed PMID: 37560735]
Level 1 (high-level) evidenceKaraca Z, Kelestimur F. The management of hypophysitis. Minerva endocrinologica. 2016 Sep:41(3):390-9 [PubMed PMID: 26963662]
Angelousi A, Alexandraki K, Tsoli M, Kaltsas G, Kassi E. Hypophysitis (Including IgG4 and Immunotherapy). Neuroendocrinology. 2020:110(9-10):822-835. doi: 10.1159/000506903. Epub 2020 Mar 4 [PubMed PMID: 32126548]
Honegger J, Buchfelder M, Schlaffer S, Droste M, Werner S, Strasburger C, Störmann S, Schopohl J, Kacheva S, Deutschbein T, Stalla G, Flitsch J, Milian M, Petersenn S, Elbelt U, Pituitary Working Group of the German Society of Endocrinology. Treatment of Primary Hypophysitis in Germany. The Journal of clinical endocrinology and metabolism. 2015 Sep:100(9):3460-9. doi: 10.1210/jc.2015-2146. Epub 2015 Jun 19 [PubMed PMID: 26091204]
Lin M, Tsang V, Brewer J, Clifton-Bligh R, Gild ML. Infiltrative lymphocytic hypophysitis successfully treated with rituximab and mycophenolate mofetil. Endocrinology, diabetes & metabolism case reports. 2020 Jul 29:2020():. pii: EDM200041. doi: 10.1530/EDM-20-0041. Epub 2020 Jul 29 [PubMed PMID: 32729848]
Level 3 (low-level) evidenceLanglois F, Varlamov EV, Fleseriu M. Hypophysitis, the Growing Spectrum of a Rare Pituitary Disease. The Journal of clinical endocrinology and metabolism. 2022 Jan 1:107(1):10-28. doi: 10.1210/clinem/dgab672. Epub [PubMed PMID: 34528683]
Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, Chari ST, Della-Torre E, Frulloni L, Goto H, Hart PA, Kamisawa T, Kawa S, Kawano M, Kim MH, Kodama Y, Kubota K, Lerch MM, Löhr M, Masaki Y, Matsui S, Mimori T, Nakamura S, Nakazawa T, Ohara H, Okazaki K, Ryu JH, Saeki T, Schleinitz N, Shimatsu A, Shimosegawa T, Takahashi H, Takahira M, Tanaka A, Topazian M, Umehara H, Webster GJ, Witzig TE, Yamamoto M, Zhang W, Chiba T, Stone JH, Second International Symposium on IgG4-Related Disease. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis & rheumatology (Hoboken, N.J.). 2015 Jul:67(7):1688-99. doi: 10.1002/art.39132. Epub [PubMed PMID: 25809420]
Level 3 (low-level) evidenceKhaleghi M, Finger G, Wu KC, Munjal V, Ghalib L, Kobalka P, Blakaj D, Dibs K, Carrau R, Prevedello D. Successful treatment of medically and surgically refractory lymphocytic hypophysitis with fractionated stereotactic radiotherapy: a single-center experience and systematic literature review. Pituitary. 2024 Apr:27(2):213-229. doi: 10.1007/s11102-023-01367-8. Epub 2024 Jan 25 [PubMed PMID: 38270722]
Level 1 (high-level) evidenceSaeger W. [Hypophysitis : Types and differential diagnosis]. Der Pathologe. 2016 May:37(3):230-7. doi: 10.1007/s00292-016-0164-x. Epub [PubMed PMID: 27103256]
Solinas C, Porcu M, De Silva P, Musi M, Aspeslagh S, Scartozzi M, Willard-Gallo K, Mariotti S, Saba L. Cancer immunotherapy-associated hypophysitis. Seminars in oncology. 2018 Jun:45(3):181-186. doi: 10.1053/j.seminoncol.2018.09.002. Epub 2018 Oct 21 [PubMed PMID: 30352754]
Kyriacou A, Gnanalingham K, Kearney T. Lymphocytic hypophysitis: modern day management with limited role for surgery. Pituitary. 2017 Apr:20(2):241-250. doi: 10.1007/s11102-016-0769-3. Epub [PubMed PMID: 27778295]
Donegan D, Saeed Z, Delivanis DA, Murad MH, Honegger J, Amereller F, Oguz SH, Erickson D, Bancos I. Outcomes of Initial Management Strategies in Patients With Autoimmune Lymphocytic Hypophysitis: A Systematic Review and Meta-analysis. The Journal of clinical endocrinology and metabolism. 2022 Mar 24:107(4):1170-1190. doi: 10.1210/clinem/dgab839. Epub [PubMed PMID: 35137155]
Level 1 (high-level) evidence