Introduction
Hyperimmunoglobulin E syndromes (HIESs) are defined by the triad of recurrent cutaneous and sinopulmonary infections, dermatitis beginning in infancy or early childhood, and markedly elevated immunoglobulin E (IgE) levels. David et al first described autosomal dominant HIES as Job syndrome in 1966 in 2 patients with eczema, recurrent pulmonary infections, and cold lung abscesses (see Image. Job Syndrome). Later, in 1972, Buckley et al reported an association between this condition and elevated serum IgE levels and a series of phenotypic features. In 2007, researchers discovered that classic autosomal dominant HIES is caused by deficiency of the signal transducer and activator of transcription 3 (STAT3) gene. Subsequently, numerous other HIES variants resulting from abnormalities in proteins involved in STAT3 signaling or other immune regulatory pathways have been recognized.[1][2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The hyper-IgE syndromes are classified into 2 types:
- Type I: Autosomal dominant hyper-IgE syndrome (AD-HIES), also known as Job syndrome or signal transducer and activator of transcription 3 (STAT3) gene–HIES, in which patients generally exhibit several nonimmunologic features, including connective tissue, dental, and skeletal abnormalities.
- Type II: Autosomal recessive hyperimmunoglobulin E syndrome (AR-HIES), also known as dedicator of cytokinesis 8 (DOCK8) gene–HIES, which has a higher incidence of neurologic complications and cutaneous viral infections.
Newer classifications include phosphoglucomutase 3 gene (PGM3), tyrosine kinase 2 gene (TYK2), zinc finger protein 341 gene (ZNF341), caspase recruitment domain family member 11 gene (CARD11), and interleukin 6 receptor gene (IL6R) deficiencies, which can produce overlapping symptoms.[4]
Epidemiology
Autosomal dominant hyper-IgE syndrome is an uncommon condition, and its actual incidence remains undetermined. The incidence of this disease exists across all racial and ethnic groups, exhibiting no apparent sex predilection. The disease typically develops in infancy, but diagnosis may be delayed until childhood or adulthood. Enhanced awareness and genetic testing have facilitated the early identification of affected families. AR-HIES, predominantly resulting from DOCK8 deficiency, is infrequent and manifests in both consanguineous and nonconsanguineous populations.
Pathophysiology
Heterozygous mutations in the signal transducer and activator of transcription 3 gene (STAT3) result in classic autosomal dominant hyperimmunoglobulin E syndrome (HIES), often known as STAT3-HIES. Various combinations of receptor-associated Janus kinases and STAT proteins transduce cytokine signals; following Janus kinase–mediated phosphorylation, the STAT proteins dimerize, translocate to the nucleus, and activate target genes. Vulnerability to bacterial and candidal infections in HIES underscores the importance of STAT3-dependent cytokines (eg, interleukin [IL]-6, IL-21, and IL-23) in the differentiation of IL-17–producing CD4+ T cells that contribute to defense against these pathogens. Insufficient stimulation of β-defensin synthesis by IL-22, produced by T helper 17 cells and signaling through STAT3, may facilitate the onset of infections at epithelial surfaces (eg, skin and lungs) in STAT3-HIES. The STAT3 protein is essential for the signaling pathways of IL-6, which facilitates acute-phase responses and functions as a pyrogen, and of IL-10, an anti-inflammatory cytokine. STAT3 deficiency explains the occurrence of cold abscesses and the damaging inflammation, such as that found in the lung, which are characteristic of HIES. Impaired production of IL-10–induced tolerogenic dendritic cells and forkhead box P3–positive regulatory T cells may exacerbate other inflammatory characteristics of HIES, including atopic-like dermatitis and elevated IgE levels.[5] STAT3 also inhibits osteoclast development, and osteoporosis is characteristic of STAT3 deficiency in both mice and patients with HIES. Finally, leukocytes from persons lacking STAT3 generate elevated levels of interferon-γ and tumor necrosis factor (TNF) when stimulated with IL-12 and lipopolysaccharide/interferon-γ, respectively, compared with leukocytes from unaffected controls.[1][2]
Additional variants of hyperimmunoglobulin E syndrome may arise from the disruption of STAT3 signaling, including deficiencies in the following proteins: the IL-6 receptor (autosomal recessive) and IL-6 signal transducer (glycoprotein 130; autosomal dominant or autosomal recessive); phosphoglucomutase 3 (PGM3; autosomal recessive), which enhances glycoprotein 130 expression; zinc finger protein 341 (ZNF341; autosomal recessive), which modulates STAT3 expression; and ERBIN (autosomal dominant), whose expression is induced by STAT3. Generally, these conditions exhibit clinical characteristics that overlap with those of STAT3-HIES, including additional manifestations such as autoimmunity and cognitive impairment in PGM3 deficiency and craniosynostosis in autosomal recessive glycoprotein 130 deficiency.[6]
DOCK8 deficiency is an autosomal recessive disorder that shares the characteristics of HIES. The DOCK8 protein associates with the Wiskott-Aldrich syndrome protein in a complex that connects T-cell receptors to the actin cytoskeleton. DOCK8 deficiency leads to increased T helper 2 cells and the synthesis of the pruritogen IL-31 while concurrently reducing T helper 1 and T helper 17 cells.[7][8][9] The caspase recruitment domain family member 11 (CARD11) gene–associated atopy with dominant interference with nuclear factor κB signaling arises from heterozygous dominant-negative mutations in CARD11, which encodes a scaffolding protein integral to lymphocyte receptor signaling. Patients exhibit the distinctive HIES triad along with additional atopic symptoms and B-cell lymphopenia. The characteristics of HIES have also been recognized in a subset of individuals with homozygous mutations in the tyrosine kinase 2 gene (TYK2), a member of the Janus kinase family. Affected patients exhibit deficiencies in the signaling pathways for IL-12 and interferon-α/β, resulting in increased vulnerability to mycobacterial and viral infections and alterations in IL-6 and IL-10 levels.[10][11]
Histopathology
The papulopustular eruption in STAT3-HIES is histologically defined by eosinophilic folliculitis, eosinophilic spongiosis, and a superficial and deep perivascular infiltration rich in eosinophils.[12][13]
History and Physical
The classic autosomal dominant form (STAT3-HIES) is characterized by clinical signs of immunological and nonimmunological features:
Immunological Features
Individuals with STAT3-HIES generally present within the first month of life with a noninfectious folliculocentric papulopustular eruption affecting the face, scalp, neck, axillae, and diaper region (see Image. Job syndrome). Chronic candidiasis of the oral mucosa, periungual regions, and nails affects approximately 80% of patients with HIES and may signify the initial infectious presentation in infancy.[13] Cutaneous staphylococcal infections involve impetiginized plaques, retroauricular fissures, folliculitis, furunculosis, abscesses, cellulitis, lymphangitis, and paronychia, which can result in nail dystrophy. Cutaneous abscesses are frequently large and predominantly affect the neck, scalp, periorbital region, axillae, and groin. These lesions are called cold abscesses because they lack the redness and tenderness typically observed in healthy individuals. Patients frequently are afebrile or exhibit only a low-grade fever. Although the abscesses are predominantly staphylococcal, many patients also experience recurrent skin infections caused by other pathogens, including Streptococcus pyogenes and Candida albicans.[13] The eczematous rash associated with HIES has numerous clinical features similar to those of atopic dermatitis, such as intense itching, lichenification, and staphylococcal superinfection. This rash is typically observed in young children but often resolves by puberty. In contrast to patients with atopic dermatitis, patients with STAT3-HIES typically do not display allergic rhinitis, asthma, or other dermatological manifestations of atopy.[13]
Although many patients with STAT3-HIES exhibit only cutaneous signs, most also experience recurrent bronchitis and pneumonia. Staphylococcus aureus and Haemophilus influenzae organisms typically cause lung infections that can lead to empyema, bronchiectasis, and pneumatocele formation. Pneumatoceles often persist and become susceptible to recurrent infections by bacterial or fungal agents, especially Aspergillus species. Occasionally, significant hemoptysis occurs. Pneumocystis jirovecii pneumonia may also manifest in newborns and children with STAT3-HIES. Additional common locations of infection include the ears, oral mucosa, sinuses, and eyes. Visceral infections, except pneumonia, are uncommon. However, deep-seated infection has been reported as a retroperitoneal abscess.[14]
Nonimmunological Features
Patients with STAT3-HIES exhibit gradual facial coarsening characterized by thickened, doughy skin, enlarged follicular ostia, pitted scarring, a broad nasal bridge, a wide, fleshy nasal tip, deep-set eyes, a prominent forehead, and irregularly proportioned cheeks and jaw. Osteopenia is frequently observed and is associated with an increased risk of fractures in the long bones, ribs, and pelvis. More than 50% of adolescents and adults with STAT3-HIES have experienced at least 3 fractures, sometimes resulting from undiagnosed or minor trauma. Scoliosis is present in 75% of patients aged 16 years or older, whereas joint hyperextensibility affects approximately 70% of patients. About 50% of patients with STAT3-HIES exhibit a high-arched palate, and distinctive dental manifestations include retention of primary teeth due to inadequate root resorption and absence of eruption of secondary teeth.
Numerous cerebral anomalies have been identified, including Chiari malformations, lacunar infarctions, and localized white matter hyperintensities on T2-weighted magnetic resonance imaging. The occurrence of coronary artery aneurysms in adults has also been documented. HIES is associated with an increased risk of B-cell-origin non–Hodgkin lymphoma.
DOCK8 deficiency resembles STAT3-HIES, characterized by markedly elevated serum IgE levels, peripheral eosinophilia, chronic eczematous dermatitis, recurrent staphylococcal skin infections (including cold abscesses), respiratory tract infections, and mucocutaneous candidiasis. However, skeletal and dental deformities, facial coarsening, and pneumatoceles are not defining characteristics. Individuals with DOCK8 deficiency are susceptible to warts, molluscum contagiosum, severe herpes simplex, and varicella-zoster virus infections, as well as opportunistic infections. Further manifestations include asthma, food allergies leading to anaphylaxis, autoimmunity, cerebral vasculitis, mucocutaneous squamous cell carcinomas, and lymphomas. Up to 50% of patients die by age 20 years due to infection, malignant neoplasms, or central nervous system disorders.[15][16]
Evaluation
Genetic testing is the optimal diagnostic approach. STAT3 sequencing confirms the presence of autosomal dominant hyper-IgE syndrome. Testing for DOCK8 deficiency should be conducted when severe viral infections, food allergies, or failure to thrive are significant characteristics. Additional uncommon etiologies include mutations of TYK2 and ZNF341.[17] Patients with HIES exhibit very high blood concentrations of polyclonal IgE, typically exceeding 2000 IU/mL, which occasionally diminish in adulthood. Patients have elevated concentrations of antistaphylococcal and anticandidal IgE antibodies and frequently experience acute wheal-and-flare responses to various dietary and environmental allergens. Numerous affected patients exhibit eosinophilia in both peripheral blood and sputum. Cell-mediated immunity is frequently abnormal, evidenced by anergy in skin testing and diminished in vitro lymphoproliferative responses to certain antigens. Serum concentrations of IgG, IgA, and IgM are often within reference ranges in STAT3-HIES, as are lymphocyte subsets. Chest radiography, chest CT, or both are essential for assessing the extent of lung parenchymal involvement in autosomal dominant HIES.[18] Conversely, DOCK8 deficiency represents a mixed immunodeficiency marked by lymphopenia, defined by a deficit of CD4+ T cells relative to CD8+ T cells and B cells, diminished IgM levels, fluctuating IgG levels, raised IgE levels, and peripheral eosinophilia.[19][20]
Treatment / Management
The management of infectious challenges in HIES involves incision and drainage of abscesses, along with both therapeutic and preventive antibiotic therapy. Interferon-γ has demonstrated efficacy in managing infections, while intravenous immunoglobulin may improve dermatitis, prevent infections, and reduce IgE levels. The administration of dupilumab and omalizumab has been shown to improve eczematous dermatitis associated with HIES.[21][22]
The treatment of nonimmunologic manifestations must be managed by an interprofessional team to prevent complications. Consequently, scoliosis, depending on its severity, as well as bone fractures and degenerative joint disease, may require orthopedic surgical procedures. Dental abnormalities require adequate stomatologic treatment. Cardiovascular complications are treated in a specialized environment.[23][24][25][26]
Bone-marrow transplant has yielded variable outcomes in these patients.[27] Hematopoietic stem cell transplantation is typically recommended for severe DOCK8-related disorders or refractory STAT3-HIES when standard treatments are inadequate. In DOCK8-HIES, hematopoietic stem cell transplant is considered curative when performed before irreversible end-organ damage, restoring T-cell and natural killer cell function and significantly reducing the infection burden.[28][29]
Differential Diagnosis
The current diagnostic criteria for STAT3-HIES require an IgE level exceeding 1000 IU/mL and a cumulative score of 5 clinical symptoms, which include pneumonia (confirmed by radiography), newborn papulopustular eruption (occurring during the first 3 weeks of life), pathologic bone fractures, distinctive facial features, and a high-arched palate. The presence of these traits, along with a deficiency of T helper 17 cells or a heterozygous STAT3 mutation, supports a probable or definitive diagnosis, respectively.[30]
Hyper-IgE syndromes must be distinguished from several other disorders that may present with elevated IgE levels and a skin eruption, including atopic dermatitis, Wiskott-Aldrich syndrome, Netherton syndrome, Omenn syndrome, DiGeorge syndrome, immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, ARPC1B (actin-related protein 2 and 3 complex subunit 1B) gene deficiency, Loeys-Dietz syndrome, and graft-versus-host disease. Atopic dermatitis and Wiskott-Aldrich disease are sometimes confused with HIES because of their eczematous dermatitis and staphylococcal infections. Nonetheless, the emergence of cold abscesses, recurrent pneumonia, pronounced facial features, and osteopenia helps distinguish STAT3-HIES from these conditions, whereas platelet anomalies help distinguish Wiskott-Aldrich syndrome and ARPC1B syndrome. Moreover, the rash typically manifests at birth or within the first few weeks, in contrast to atopic dermatitis, which typically emerges after age 3 months. Flow cytometry would also reveal a notable decrease in T helper 17 cells in STAT3-HIES and an excess of T helper 2 cells in atopic dermatitis.
Patients with prolidase deficiency exhibit elevated IgE levels, susceptibility to pyogenic infections, and eczematous dermatitis, alongside recurrent leg ulcers, facial dysmorphism, and intellectual disability. Patients with chronic granulomatous disease and myeloperoxidase deficiency develop bacterial and candidal abscesses; nevertheless, they generally do not have significantly elevated IgE levels. CD4+ lymphocytopenia may be a prominent feature of DOCK8 deficiency and other immunodeficiency disorders associated with a predisposition to verruca formation, including epidermodysplasia verruciformis and Netherton syndrome.
Prognosis
Patients with AD-HIES experience a shortened lifespan, with mortality primarily occurring in adulthood due to chronic lung infections. Historically, within a specific cohort, the mean age of surviving patients was 27 years, while infection-related mortality occurred, on average, at 29 years.[31] Mortality is typically associated with complications from pneumatoceles or bronchiectasis, including secondary infections by molds such as Aspergillus or Scedosporium species, which can result in vascular invasion, metastatic dissemination, or fatal hemoptysis. Patients also face an elevated risk for hematologic malignancies, such as Hodgkin and non–Hodgkin lymphoma.[32] Although autosomal dominant–HIES is associated with high morbidity and mortality, advances in medical care and close monitoring have led to a recent improvement in prognosis.[23][33] Conversely, autosomal recessive hyper-IgE syndrome resulting from DOCK8 deficiency is linked to markedly increased mortality in the absence of hematopoietic stem cell transplant. Autosomal recessive HIES is also associated with more deaths due to sepsis, central nervous system infections, and vasculitis than autosomal dominant HIES.[34]
Complications
Complications stem from recurrent infections, anatomic anomalies, and immunologic dysregulation, and pulmonary disease is the primary cause of morbidity and mortality. Other complications include:
- Malignant neoplasms, especially non–Hodgkin lymphoma. Other cancers have also been reported, such as Hodgkin lymphoma and cancers of the vulva and lung
- Autoimmune diseases like systemic lupus erythematosus, membranoproliferative glomerulonephritis, vasculitis, and dermatomyositis
- Hypertension associated with vascular abnormalities
- Myocardial infarction due to the rupture of coronary aneurysms
- Lacunar infarcts due to the rupture of cerebral aneurysms [27][35]
Deterrence and Patient Education
Patients should maintain follow-up with an immunologist and an interprofessional care team because the disorder affects multiple organ systems. Maintaining general hygiene is vital for reducing the risk of infection.
Pearls and Other Issues
Dominant-negative mutations in the STAT3 gene are the primary etiologic cause of HIES. The autosomal recessive form of HIES is caused by loss-of-function mutations in the DOCK8 gene. STAT3 binds to the promoters of genes, including those involved in cytokine signaling, leukemia inhibitory factor, corticotropin, oncostatin, receptor tyrosine kinases, insulin-like growth factor 1, and growth hormone. Therefore, STAT3 is critical for cell survival and inflammation in the skin, lungs, thymus, mammary glands, macrophages, neurons, and lymphocytes. Mutations affecting hepatic IL-6 responses alter acute-phase protein synthesis.[36] The condition is characterized by defects in immunity with eczematous and nonimmunologic systemic disorders. Antenatal diagnosis is theoretically possible. However, the very low incidence and sporadic nature in most cases make this method of little interest. Current treatment is purely symptomatic. Early detection and treatment of infections are essential for disease treatment. Exposure to chlorinated water or dilute bleach baths can diminish staphylococcal skin colonization. Bacillus Calmette-Guérin vaccination is contraindicated in patients with known or suspected immunologic deficits, and live vaccines should be deferred until age 6 months.[37] Immunology research has led to the discovery of new treatment options and improved understanding of HIES. The addition of omalizumab to standard treatment may improve pulmonary and dermatologic symptoms.[38] Further research is needed to improve HIES diagnosis and care, as substantial gaps remain in our understanding of the disorder.[39]
Enhancing Healthcare Team Outcomes
The hyper-IgE syndromes are rare immunologic disorders characterized by recurrent infections. Early identification and treatment of patients with these syndromes are imperative for reducing morbidity and mortality. The care of patients with these conditions requires collaboration among healthcare professionals to ensure patient-centered care and improve overall outcomes. The conditions are primarily treated by hematologists and infectious disease specialists, whereas outpatient follow-up is conducted by primary care clinicians and nurse practitioners as part of interprofessional team care. Most of these patients require long-term antibiotic therapy combined with abscess drainage, so pharmacist consultation is necessary to appropriately target antimicrobial therapy. The treatment of nonimmunologic manifestations must be interprofessional to treat and prevent complications. Consequently, scoliosis, depending on its severity, as well as bone fractures and degenerative joint disease, may require orthopedic surgical procedures. Dental abnormalities require adequate stomatologic treatment. Cardiovascular complications are treated in a specialized environment. An outpatient nurse should make regular home visits to ensure the patient's health remains stable and report findings to the healthcare team.
A strategic approach is equally crucial and should include evidence-based strategies to optimize treatment plans and minimize adverse effects. Ethical considerations must guide decision-making to ensure informed consent and respect patient autonomy in treatment choices. Each healthcare professional must be aware of their responsibilities and contribute specific expertise to the patient's care plan, fostering an interprofessional approach. Effective interprofessional communication is paramount, allowing seamless information exchange and collaborative decision-making among team members. Care coordination plays a pivotal role in ensuring that the patient's journey from diagnosis to treatment and follow-up is well coordinated, minimizing errors and enhancing patient safety. By embracing these principles of skill, strategy, ethics, responsibilities, interprofessional communication, and care coordination, healthcare professionals can deliver patient-centered care, ultimately improving patient outcomes and enhancing team performance in the treatment of hyper-IgE syndromes.
Media
(Click Image to Enlarge)
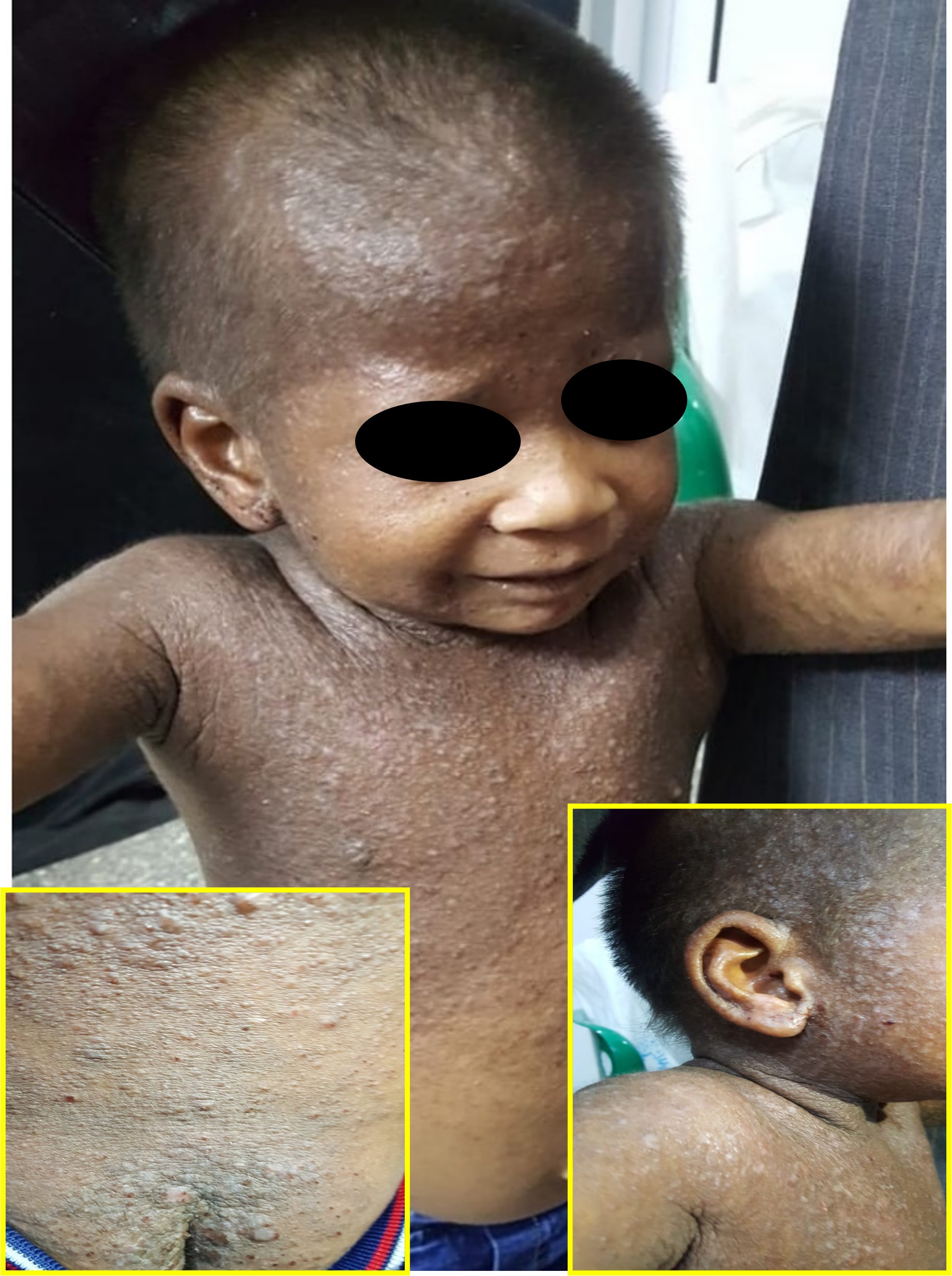
Job Syndrome. A 5-year-old child with Job syndrome manifesting in the form of widespread atopic dermatitis-like lesions, recurrent chest infections, stunted growth, and elevated IgE levels.
Contributed by HM Saleh, MD, PhD
References
Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schäffer AA, Puck JM, Grimbacher B. STAT3 mutations in the hyper-IgE syndrome. The New England journal of medicine. 2007 Oct 18:357(16):1608-19 [PubMed PMID: 17881745]
Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007 Aug 30:448(7157):1058-62 [PubMed PMID: 17676033]
Fadil I, Ben-Ali M, Jeddane L, Barbouche MR, Bousfiha AA. The Seven STAT3-Related Hyper-IgE Syndromes. Journal of clinical immunology. 2021 Aug:41(6):1384-1389. doi: 10.1007/s10875-021-01041-3. Epub 2021 Apr 26 [PubMed PMID: 33903995]
AlYafie R, Velayutham D, van Panhuys N, Jithesh PV. The genetics of hyper IgE syndromes. Frontiers in immunology. 2025:16():1516068. doi: 10.3389/fimmu.2025.1516068. Epub 2025 Feb 18 [PubMed PMID: 40040707]
Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, Yamada M, Kawamura N, Ariga T, Tsuge I, Nonoyama S, Karasuyama H, Minegishi Y. Defective IL-10 signaling in hyper-IgE syndrome results in impaired generation of tolerogenic dendritic cells and induced regulatory T cells. The Journal of experimental medicine. 2011 Feb 14:208(2):235-49. doi: 10.1084/jem.20100799. Epub 2011 Feb 7 [PubMed PMID: 21300911]
Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, Jing H, Kim ES, Biancalana M, Wolfe LA, DiMaggio T, Matthews HF, Kranick SM, Stone KD, Holland SM, Reich DS, Hughes JD, Mehmet H, McElwee J, Freeman AF, Freeze HH, Su HC, Milner JD. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. The Journal of allergy and clinical immunology. 2014 May:133(5):1400-9, 1409.e1-5. doi: 10.1016/j.jaci.2014.02.013. Epub 2014 Feb 28 [PubMed PMID: 24589341]
Yamamura K, Uruno T, Shiraishi A, Tanaka Y, Ushijima M, Nakahara T, Watanabe M, Kido-Nakahara M, Tsuge I, Furue M, Fukui Y. The transcription factor EPAS1 links DOCK8 deficiency to atopic skin inflammation via IL-31 induction. Nature communications. 2017 Jan 9:8():13946. doi: 10.1038/ncomms13946. Epub 2017 Jan 9 [PubMed PMID: 28067314]
Tangye SG, Pillay B, Randall KL, Avery DT, Phan TG, Gray P, Ziegler JB, Smart JM, Peake J, Arkwright PD, Hambleton S, Orange J, Goodnow CC, Uzel G, Casanova JL, Lugo Reyes SO, Freeman AF, Su HC, Ma CS. Dedicator of cytokinesis 8-deficient CD4(+) T cells are biased to a T(H)2 effector fate at the expense of T(H)1 and T(H)17 cells. The Journal of allergy and clinical immunology. 2017 Mar:139(3):933-949. doi: 10.1016/j.jaci.2016.07.016. Epub 2016 Aug 20 [PubMed PMID: 27554822]
Janssen E, Tohme M, Hedayat M, Leick M, Kumari S, Ramesh N, Massaad MJ, Ullas S, Azcutia V, Goodnow CC, Randall KL, Qiao Q, Wu H, Al-Herz W, Cox D, Hartwig J, Irvine DJ, Luscinskas FW, Geha RS. A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. The Journal of clinical investigation. 2016 Oct 3:126(10):3837-3851. doi: 10.1172/JCI85774. Epub 2016 Sep 6 [PubMed PMID: 27599296]
Level 2 (mid-level) evidenceMa CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, Reynolds PR, Lyons JJ, Nelson CG, Ruffo E, Dorjbal B, Glauzy S, Yamakawa N, Arjunaraja S, Voss K, Stoddard J, Niemela J, Zhang Y, Rosenzweig SD, McElwee JJ, DiMaggio T, Matthews HF, Jones N, Stone KD, Palma A, Oleastro M, Prieto E, Bernasconi AR, Dubra G, Danielian S, Zaiat J, Marti MA, Kim B, Cooper MA, Romberg N, Meffre E, Gelfand EW, Snow AL, Milner JD. Germline hypomorphic CARD11 mutations in severe atopic disease. Nature genetics. 2017 Aug:49(8):1192-1201. doi: 10.1038/ng.3898. Epub 2017 Jun 19 [PubMed PMID: 28628108]
Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, Takada H, Hara T, Kawamura N, Ariga T, Kaneko H, Kondo N, Tsuge I, Yachie A, Sakiyama Y, Iwata T, Bessho F, Ohishi T, Joh K, Imai K, Kogawa K, Shinohara M, Fujieda M, Wakiguchi H, Pasic S, Abinun M, Ochs HD, Renner ED, Jansson A, Belohradsky BH, Metin A, Shimizu N, Mizutani S, Miyawaki T, Nonoyama S, Karasuyama H. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006 Nov:25(5):745-55 [PubMed PMID: 17088085]
Chamlin SL, McCalmont TH, Cunningham BB, Esterly NB, Lai CH, Mallory SB, Mancini AJ, Tamburro J, Frieden IJ. Cutaneous manifestations of hyper-IgE syndrome in infants and children. The Journal of pediatrics. 2002 Oct:141(4):572-5 [PubMed PMID: 12378200]
Eberting CL, Davis J, Puck JM, Holland SM, Turner ML. Dermatitis and the newborn rash of hyper-IgE syndrome. Archives of dermatology. 2004 Sep:140(9):1119-25 [PubMed PMID: 15381553]
Sugiyama H, Higuchi Y, Fujiwara S, Aya K, Mukai W, Goto T. STAT3-Mutated Hyper-IgE Syndrome With Retroperitoneal Abscess in Adolescence. Clinical case reports. 2026 Feb:14(2):e71980. doi: 10.1002/ccr3.71980. Epub 2026 Feb 4 [PubMed PMID: 41659947]
Level 3 (low-level) evidenceEngelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, Saghafi S, Pourpak Z, Ceja R, Sassi A, Graham LE, Massaad MJ, Mellouli F, Ben-Mustapha I, Khemiri M, Kilic SS, Etzioni A, Freeman AF, Thiel J, Schulze I, Al-Herz W, Metin A, Sanal Ö, Tezcan I, Yeganeh M, Niehues T, Dueckers G, Weinspach S, Patiroglu T, Unal E, Dasouki M, Yilmaz M, Genel F, Aytekin C, Kutukculer N, Somer A, Kilic M, Reisli I, Camcioglu Y, Gennery AR, Cant AJ, Jones A, Gaspar BH, Arkwright PD, Pietrogrande MC, Baz Z, Al-Tamemi S, Lougaris V, Lefranc G, Megarbane A, Boutros J, Galal N, Bejaoui M, Barbouche MR, Geha RS, Chatila TA, Grimbacher B. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. The Journal of allergy and clinical immunology. 2015 Aug:136(2):402-12. doi: 10.1016/j.jaci.2014.12.1945. Epub 2015 Feb 25 [PubMed PMID: 25724123]
Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, Kostyuchenko L, Genel F, Kütükcüler N, Karaca N, Gonzalez-Granado L, Abbott J, Al-Zahrani D, Rezaei N, Baz Z, Thiel J, Ehl S, Marodi L, Orange JS, Sawalle-Belohradsky J, Keles S, Holland SM, Sanal Ö, Ayvaz DC, Tezcan I, Al-Mousa H, Alsum Z, Hawwari A, Metin A, Matthes-Martin S, Hönig M, Schulz A, Picard C, Barlogis V, Gennery A, Ifversen M, van Montfrans J, Kuijpers T, Bredius R, Dückers G, Al-Herz W, Pai SY, Geha R, Notheis G, Schwarze CP, Tavil B, Azik F, Bienemann K, Grimbacher B, Heinz V, Gaspar HB, Aydin R, Hagl B, Gathmann B, Belohradsky BH, Ochs HD, Chatila T, Renner ED, Su H, Freeman AF, Engelhardt K, Albert MH, inborn errors working party of EBMT. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. Journal of clinical immunology. 2015 Feb:35(2):189-98. doi: 10.1007/s10875-014-0126-0. Epub 2015 Jan 28 [PubMed PMID: 25627830]
Tsilifis C, Freeman AF, Gennery AR. STAT3 Hyper-IgE Syndrome-an Update and Unanswered Questions. Journal of clinical immunology. 2021 Jul:41(5):864-880. doi: 10.1007/s10875-021-01051-1. Epub 2021 May 1 [PubMed PMID: 33932191]
Jończyk-Potoczna K, Szczawińska-Popłonyk A, Warzywoda M, Bręborowicz A, Pawlak B. Hyper Ig E syndrome (Job syndrome, HIES) - radiological images of pulmonary complications on the basis of three cases. Polish journal of radiology. 2012 Apr:77(2):69-72 [PubMed PMID: 22844313]
Level 3 (low-level) evidenceZhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, Matthews HF, Davis J, Turner ML, Uzel G, Holland SM, Su HC. Combined immunodeficiency associated with DOCK8 mutations. The New England journal of medicine. 2009 Nov 19:361(21):2046-55. doi: 10.1056/NEJMoa0905506. Epub 2009 Sep 23 [PubMed PMID: 19776401]
Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, Chen A, Kim HS, Lloret MG, Schulze I, Ehl S, Thiel J, Pfeifer D, Veelken H, Niehues T, Siepermann K, Weinspach S, Reisli I, Keles S, Genel F, Kutukculer N, Camcioğlu Y, Somer A, Karakoc-Aydiner E, Barlan I, Gennery A, Metin A, Degerliyurt A, Pietrogrande MC, Yeganeh M, Baz Z, Al-Tamemi S, Klein C, Puck JM, Holland SM, McCabe ER, Grimbacher B, Chatila TA. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2009 Dec:124(6):1289-302.e4. doi: 10.1016/j.jaci.2009.10.038. Epub [PubMed PMID: 20004785]
Staudacher O, Krüger R, Kölsch U, Thee S, Gratopp A, Wahn V, Lau S, von Bernuth H. Relieving job: Dupilumab in autosomal dominant STAT3 hyper-IgE syndrome. The journal of allergy and clinical immunology. In practice. 2022 Jan:10(1):349-351.e1. doi: 10.1016/j.jaip.2021.08.042. Epub 2021 Sep 16 [PubMed PMID: 34536614]
Lu CW, Lee WI, Chung WH. Dupilumab for STAT3-Hyper-IgE Syndrome With Refractory Intestinal Complication. Pediatrics. 2021 Sep:148(3):. pii: e2021050351. doi: 10.1542/peds.2021-050351. Epub 2021 Aug 20 [PubMed PMID: 34417287]
He YY, Liu B, Xiao XP. [Hyper-IgE syndromes]. Lin chuang er bi yan hou tou jing wai ke za zhi = Journal of clinical otorhinolaryngology head and neck surgery. 2017 Jun 5:31(11):892-896. doi: 10.13201/j.issn.1001-1781.2017.11.019. Epub [PubMed PMID: 29775011]
Santana PRP, Medeiros AK, Barbisan CC, Gomes ACP, Marchiori E. Hyperimmunoglobulin E syndrome (Job syndrome): chest CT findings. Jornal brasileiro de pneumologia : publicacao oficial da Sociedade Brasileira de Pneumologia e Tisilogia. 2018 Jul-Aug:44(4):335-336. doi: 10.1590/S1806-37562017000000175. Epub [PubMed PMID: 30020346]
Peña-López S, Monteagudo B, Fernández-Jorge B, García-Fernández ME. [Cutaneous manifestations in an infant with hyper-IgE syndrome]. Anales de pediatria. 2019 May:90(5):319-320. doi: 10.1016/j.anpedi.2018.03.015. Epub 2018 Apr 24 [PubMed PMID: 29703558]
Sharma A, Kumar S, Jagia P. Pulmonary Artery Pseudoaneurysm in Hyper-IgE Syndrome: Rare Complication With Successful Endovascular Management. Vascular and endovascular surgery. 2018 Jul:52(5):375-377. doi: 10.1177/1538574418762656. Epub 2018 Mar 18 [PubMed PMID: 29552943]
Yong PF, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B. An update on the hyper-IgE syndromes. Arthritis research & therapy. 2012 Nov 30:14(6):228. doi: 10.1186/ar4069. Epub 2012 Nov 30 [PubMed PMID: 23210525]
Harrison SC, Tsilifis C, Slatter MA, Nademi Z, Worth A, Veys P, Ponsford MJ, Jolles S, Al-Herz W, Flood T, Cant AJ, Doffinger R, Barcenas-Morales G, Carpenter B, Hough R, Haraldsson Á, Heimall J, Grimbacher B, Abinun M, Gennery AR. Hematopoietic Stem Cell Transplantation Resolves the Immune Deficit Associated with STAT3-Dominant-Negative Hyper-IgE Syndrome. Journal of clinical immunology. 2021 Jul:41(5):934-943. doi: 10.1007/s10875-021-00971-2. Epub 2021 Feb 1 [PubMed PMID: 33523338]
Aydin SE, Freeman AF, Al-Herz W, Al-Mousa HA, Arnaout RK, Aydin RC, Barlogis V, Belohradsky BH, Bonfim C, Bredius RG, Chu JI, Ciocarlie OC, Doğu F, Gaspar HB, Geha RS, Gennery AR, Hauck F, Hawwari A, Hickstein DD, Hoenig M, Ikinciogullari A, Klein C, Kumar A, Ifversen MRS, Matthes S, Metin A, Neven B, Pai SY, Parikh SH, Picard C, Renner ED, Sanal Ö, Schulz AS, Schuster F, Shah NN, Shereck EB, Slatter MA, Su HC, van Montfrans J, Woessmann W, Ziegler JB, Albert MH, Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation and the European Society for Primary Immunodeficiencies. Hematopoietic Stem Cell Transplantation as Treatment for Patients with DOCK8 Deficiency. The journal of allergy and clinical immunology. In practice. 2019 Mar:7(3):848-855. doi: 10.1016/j.jaip.2018.10.035. Epub 2018 Nov 2 [PubMed PMID: 30391550]
Woellner C, Gertz EM, Schäffer AA, Lagos M, Perro M, Glocker EO, Pietrogrande MC, Cossu F, Franco JL, Matamoros N, Pietrucha B, Heropolitańska-Pliszka E, Yeganeh M, Moin M, Español T, Ehl S, Gennery AR, Abinun M, Breborowicz A, Niehues T, Kilic SS, Junker A, Turvey SE, Plebani A, Sánchez B, Garty BZ, Pignata C, Cancrini C, Litzman J, Sanal O, Baumann U, Bacchetta R, Hsu AP, Davis JN, Hammarström L, Davies EG, Eren E, Arkwright PD, Moilanen JS, Viemann D, Khan S, Maródi L, Cant AJ, Freeman AF, Puck JM, Holland SM, Grimbacher B. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2010 Feb:125(2):424-432.e8. doi: 10.1016/j.jaci.2009.10.059. Epub [PubMed PMID: 20159255]
Freeman AF, Kleiner DE, Nadiminti H, Davis J, Quezado M, Anderson V, Puck JM, Holland SM. Causes of death in hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2007 May:119(5):1234-40 [PubMed PMID: 17335882]
Leonard GD, Posadas E, Herrmann PC, Anderson VL, Jaffe ES, Holland SM, Wilson WH. Non-Hodgkin's lymphoma in Job's syndrome: a case report and literature review. Leukemia & lymphoma. 2004 Dec:45(12):2521-5 [PubMed PMID: 15621772]
Level 3 (low-level) evidenceBazregari S, Azizi G, Tavakol M, Asgardoon MH, Kiaee F, Tavakolinia N, Valizadeh A, Abolhassani H, Aghamohammadi A. Evaluation of infectious and non-infectious complications in patients with primary immunodeficiency. Central-European journal of immunology. 2017:42(4):336-341. doi: 10.5114/ceji.2017.72825. Epub 2017 Dec 30 [PubMed PMID: 29479289]
Freeman AF, Holland SM. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatric research. 2009 May:65(5 Pt 2):32R-37R. doi: 10.1203/PDR.0b013e31819dc8c5. Epub [PubMed PMID: 19190525]
Szczawinska-Poplonyk A, Kycler Z, Pietrucha B, Heropolitanska-Pliszka E, Breborowicz A, Gerreth K. The hyperimmunoglobulin E syndrome--clinical manifestation diversity in primary immune deficiency. Orphanet journal of rare diseases. 2011 Nov 15:6():76. doi: 10.1186/1750-1172-6-76. Epub 2011 Nov 15 [PubMed PMID: 22085750]
Minegishi Y. Hyper-IgE syndrome, 2021 update. Allergology international : official journal of the Japanese Society of Allergology. 2021 Oct:70(4):407-414. doi: 10.1016/j.alit.2021.07.007. Epub 2021 Aug 18 [PubMed PMID: 34419355]
Staines-Boone AT, Venegas-Montoya E, García Campos JA, Obregón García JA, Marmolejo Bjirsdorp JC, Salazar Gálvez Y, Cruz-Muñoz ME, Blancas Galicia L, Medina-Torres EA, Espinosa-Padilla SE, Olguín Calderón D, Berrón-Ruiz L, Lugo Reyes SO. Job's not done: BCG-itis as the first manifestation of hyper-IgE syndrome. A case report and review of the literature. Clinical immunology (Orlando, Fla.). 2026 Jan:282():110639. doi: 10.1016/j.clim.2025.110639. Epub 2025 Nov 14 [PubMed PMID: 41242409]
Level 3 (low-level) evidenceLi H, Wang Y, Tao Y, Wu S, Xu Q, Lu C, Huang Z, Zhai Y, Chen D. Efficacy of omalizumab in the treatment of STAT3 loss-of-function mutations associated with autosomal dominant hyperimmunoglobulin E syndrome-a case report. Translational pediatrics. 2025 Nov 30:14(11):3204-3212. doi: 10.21037/tp-2025-379. Epub 2025 Nov 25 [PubMed PMID: 41367532]
Level 3 (low-level) evidenceDave T, Tashrifwala FAA, Rangwala US, Hameed R. Hyper-IgE syndrome: a case report. Annals of medicine and surgery (2012). 2024 Feb:86(2):1205-1209. doi: 10.1097/MS9.0000000000001670. Epub 2024 Jan 3 [PubMed PMID: 38333292]
Level 3 (low-level) evidence