Introduction
Interstitial lung disease (ILD), also known as diffuse parenchymal lung diseases, refers to a heterogeneous group of distinct lung disorders characterized by shared clinical, radiographic, physiologic, or pathologic features. The pathogenesis sequence involves a series of inflammation and fibrosis that extends beyond disrupting the interstitial bed (as the name implies) to changing the parenchyma (alveoli, alveolar ducts, and bronchioles). The pattern of disease spread varies among the groups; therefore, establishing the correct diagnosis is crucial.
ILD collectively represents a significant cause of morbidity and mortality, with idiopathic pulmonary fibrosis carrying a median survival of 2 to 5 years from diagnosis, underscoring the importance of early recognition and accurate subtype classification.[1] This resource focuses on the classification of ILD, etiology, epidemiology, clinical presentation, and approach to management in adults. Other specific topics related to particular causes, such as interstitial pulmonary fibrosis, idiopathic pulmonary fibrosis (see Image. Idiopathic Pulmonary Fibrosis), collagen vascular disorders, or nonspecific interstitial pneumonitis, will be discussed separately. Please see StatPearls' companion references, "Interstitial Pulmonary Fibrosis," "Idiopathic Pulmonary Fibrosis," "Nonspecific Interstitial Pneumonia," and "Collagen Vascular Disease" for further information.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The classification system used to describe ILD categorizes conditions based on clinical, histopathological, or radiologic parameters.[2] Clinical classification groups ILD by its causes to help differentiate exogenous or endogenous factors.[3] ILD of unknown origin is classified as idiopathic (primary) based on histopathological and radiological assessments. This classification encompasses conditions such as sarcoidosis, cryptogenic organizing pneumonia, and idiopathic interstitial pneumonia, which includes desquamative interstitial pneumonia (or newly named alveolar macrophage pneumonia [AMP]), idiopathic pulmonary fibrosis (IPF), nonspecific interstitial pneumonitis, acute interstitial pneumonia, and idiopathic interstitial pneumonia with autoimmune features.[4] Please see StatPearls' companion references, "Desquamative Interstitial Pneumonia," "Idiopathic Interstitial Pneumonia With Autoimmune Features," "Idiopathic Pulmonary Fibrosis," and "Acute Interstitial Pneumonia," for further information. Among these, idiopathic interstitial pneumonia is the most common, characterized by a combination of inflammation and fibrosis, which distinguishes it from infectious pneumonia.[5]
Several distinct types exist, each identified by specific histopathological features and clear clinical distinctions. Although most cases occur sporadically, genetic factors may play a role in their development. Identified etiological factors include environmental and occupational exposures. Prolonged exposure to occupational or environmental agents can exert toxic effects on the pulmonary system, with common agents including mineral dust, organic dust, and toxic gases. Various types of mineral dust are associated with ILD, with silica, asbestos, coal mine dust, beryllium, and hard metal being frequently implicated. Organic dust sources include mold spores and aerosolized bird droppings.
Inhaled toxic gases, such as methane and cyanide, affect the airways either directly or through reactive oxygen species. The extent of exposure-related injuries is challenging to quantify and likely occurs more frequently than estimated. Therefore, clinicians must conduct a comprehensive review of a patient's complete employment history and home environment to identify any potential agent-disease relationships (see Image. Etiologies of Interstitial Lung Disease).
Hypersensitivity pneumonitis results from immune-mediated lung injury following repeated inhalation of organic antigens, including avian proteins (bird fancier lung), fungal spores (farmer lung, mold-related hypersensitivity pneumonitis), and occupational agents. Identification and avoidance of the causative antigen are cornerstones of management and may halt disease progression.[6][7] Another recognized etiology of ILD is linked to connective tissue diseases, including rheumatoid arthritis, polymyositis/dermatomyositis, systemic lupus erythematosus, scleroderma, and mixed connective tissue disease. Connective tissue diseases and vasculitides impact all regions of the lungs, including the bronchioles, parenchyma, and alveoli. Consequently, ILD is a prevalent characteristic of rheumatologic conditions.[8][9]
Drug-induced ILD is a crucial factor to consider during the initial assessment of any patient with ILD. More than 350 pharmaceutical agents have been identified as potential contributors to pulmonary complications, either through the generation of reactive metabolites or as part of a broader physiological response.[10][11] Furthermore, genetic predisposition plays a role in a subset of ILD cases. Familial pulmonary fibrosis, defined as IPF in 2 or more first-degree relatives, accounts for up to 20% of IPF cases, with mutations in telomere-related genes (TERT, TERC) and surfactant protein genes (SFTPC, SFTPA2) being most commonly implicated.[12]
Epidemiology
ILD incidence rates in the United States have been difficult to determine. The reported prevalence may be low because the disease may not be recognized. ILD is a diagnosis of exclusion that requires extensive investigation.[13] ILD affects over half a million individuals and results in approximately 25,000 to 30,000 deaths annually. IPF is the most prevalent form of ILD, accounting for roughly one-third of all ILD cases, followed by connective tissue disease-related ILD, which affects approximately 1 in 4 individuals with ILD (25%). Hypersensitivity pneumonitis constitutes 15% of ILD cases.[14]
The global age-standardized prevalence of ILD is approximately 58 cases per 100,000 population, reflecting a substantial health concern worldwide.[15] Marked geographic variation exists in both the incidence of ILD and the dominant subtypes observed, influenced by regional environmental exposures, genetic predispositions, and differences in healthcare infrastructure. Enhanced recognition of ILD has been driven by advances in imaging technologies and the demographic shift toward aging populations, who are more susceptible to these conditions.
Common ILD subtypes include IPF, connective tissue disease-associated ILD (CTD-ILD), hypersensitivity pneumonitis, and sarcoidosis, each contributing to significant morbidity, mortality, and economic burden globally. However, data remain limited from resource-limited countries, where diagnostic challenges and resource constraints hinder accurate assessment and management. Results from research conducted in the United States indicate that the estimated annual incidence of IPF ranges from 6.8 to 8.8 per 100,000 individuals and from 16.3 to 17.4 per 100,000 individuals.
Additionally, the prevalence is reported to be between 14 and 27.9 cases per 100,000 individuals, and between 42.7 and 63 cases per 100,000 individuals, respectively.[16] For non-IPF ILD subtypes, an even wider range of estimated incidence and prevalence exists across regions.[17] This variation is likely due to the absence of standardized diagnostic criteria, the diverse clinical presentations and phenotypes, and differences in genetic, occupational, and other environmental factors. Sarcoidosis is the most common non-IPF ILD, particularly among those younger than 45 years of age. The estimated incidence of sarcoidosis ranges from 0.13 to 17.8 per 100,000 person-years, while its prevalence spans from 2 to 160 per 100,000 individuals. Table 1 summarizes the incidence and prevalence of the various subtypes.
Table 1. Epidemiology Estimates of Major ILD Subtypes
| ILD Subtype and Context | Incidence and Prevalence Estimate |
| Sarcoidosis (non-IPF ILD) | Incidence: approximately 0.13 to 17.8 per 100,000 person-years; Prevalence: approximately 2 to 160 per 100,000 |
| ILD associated with connective tissue disease (CTD-ILD): pooled meta-analysis across CTD subtypes | Prevalence among patients with CTD: 56% in mixed CTD, 47% in SSc, 41% in IIM, 17% in pSjD, 11% in RA, 6% in SLE |
| Progressive fibrosing ILDs (non-IPF), US (claims database) | Prevalence of fibrosing ILD: approximately 117.8 per 100,000; incidence of fibrosing ILD: approximately 51.6 per 100,000 patient-years; for progressive fibrosing phenotype, incidence approximately 32.6 per 100,000 patient-years |
| IPF prevalence | IPF incidence: 6.8–8.8 and 16.3–17.4 per 100,000 individuals.[16][18][19] |
CTD, connective tissue disease; ILD, interstitial lung disease; IIM, idiopathic inflammatory myopathies; IPF, idiopathic pulmonary fibrosis; pSjD, primary Sjögren disease; RA, rheumatoid arthritis; SSc, systemic sclerosis; SLE, systemic lupus erythematosus
Systemic sclerosis ILD is the most clinically significant CTD-ILD subtype due to its high prevalence (up to 70% to 90% on high-resolution computed tomography), association with pulmonary hypertension, and poor prognosis. All patients with systemic sclerosis should undergo baseline high-resolution computed tomography and pulmonary function testing screening regardless of respiratory symptoms.[20] Moreover, the evolution and increasing standardization of diagnostic criteria over recent decades have markedly affected the reported prevalence and incidence rates.
Enhanced imaging techniques and improved clinical awareness have led to better detection and classification of ILD subtypes, which vary geographically and epidemiologically. These findings underscore the critical need for consistent and standardized diagnostic approaches to ensure accurate epidemiological assessments, facilitate comparability across studies, and guide effective public health interventions. Taken together, these factors highlight the multifaceted nature of ILD risk, emphasizing the importance of integrated strategies that address environmental control, genetic research, and diagnostic refinement to mitigate the disease's global impact.
Pathophysiology
Many subsets of ILD have an unknown etiology. Key risk factors and drivers of ILD development encompass a range of environmental, occupational, genetic, and demographic elements.[15] Notably, exposure to environmental and occupational agents, such as dusts, fumes, and other airborne irritants, significantly contributes to the disease burden. Smoking and ambient air pollution further amplify this risk, collectively accounting for a substantial portion of the attributable risk associated with ILD. Genetic susceptibility also plays a crucial role by influencing individual vulnerability to these external factors, while the natural aging of populations exacerbates disease pathogenesis, because older age is linked to increased incidence and severity of ILD.[15]
The morphological changes seen histologically result from a sequence of inflammatory events within the parenchyma, the portion of the lung involved in gas exchange (the alveoli, alveolar ducts, and bronchioles). This compartment is the habitat for various proteins and profibrotic elements. These proteins, after repeated cycles of activation, give rise to the accumulation of connective tissue.[21] The trigger can be a known agent deposited in the lung tissue. In some cases, the fibrosis arises spontaneously. Table 2 summarizes the key pathophysiological findings in classes of ILD.
Table 2. Key Pathophysiological Findings in Classes of Interstitial Lung Disease
| Interstitial Lung Disease Category | Primary Pathophysiologic Mechanisms | Cellular Molecular Pathways | Representative Diseases |
| Idiopathic pulmonary fibrosis (IPF)[22][23] | Repetitive micro-injury to alveolar epithelium → abnormal tissue repair → fibroblast proliferation & extracellular matrix accumulation → progressive fibrosis | Transforming growth factor (TGF)-β, platelet-derived growth factor, fibroblast growth factor signaling; epithelial-mesenchymal transition; MUC5B promoter variant; telomere dysfunction; aberrant macrophage activation | Usual interstitial pneumonia (UIP) pattern, a specific scarring appearance on high-resolution computed tomography scans, characterized by basal, subpleural honeycombing, reticulation, and traction bronchiectasis [24] |
| Connective tissue disease–associated ILD (CTD-ILD)[25][26] | Autoimmune inflammation targeting alveoli, small airways, and interstitium → chronic immune activation → fibro-inflammatory remodeling | Autoantibodies (antinuclear antibody, rheumatoid factor, anti-Scl-70, antisynthetase antibodies); Th1 and Th17 pathways; immune complex deposition; macrophage-driven fibrosis | Systemic sclerosis (SSc) ILD, rheumatoid arthritis-associated ILD, Sjögren disease ILD, polymyositis and dermatomyositis ILD, mixed connective tissue disease-ILD [27] |
| Hypersensitivity pneumonitis [28] | Immune-mediated injury after inhaled antigen exposure → acute immune complex response + chronic T-cell–mediated inflammation → bronchiolocentric fibrosis when persistent | Immune complex (type III) and delayed-type hypersensitivity (type IV); CD8+ lymphocytes; granulomatous inflammation; alveolitis | Bird-fancier lung, mold-related hypersensitivity pneumonitis, farmer lung [29] |
| Occupational and environmental ILD [30] | Chronic inhalation of mineral dusts or toxic agents → direct alveolar macrophage activation → fibrosis | Silica: inflammasome activation (NLRP3), TNF-α; asbestos: ferruginous body formation, reactive oxygen species injury; coal dust: macrophage overload | Silicosis, asbestosis, and coal worker pneumoconiosis [31] |
| Drug-induced ILD [32] | Direct cytotoxicity or immune-mediated lung injury from medications → diffuse alveolar damage, inflammation, or fibrosis | Tumor necrosis factor (TNF)-α, oxidative stress, T-cell–mediated hypersensitivity; mitochondrial injury; endothelial damage | Amiodarone, methotrexate, checkpoint inhibitors, nitrofurantoin |
| Granulomatous ILD (sarcoid and nonsarcoid)[33] | Granulomatous immune reaction to unknown antigen → noncaseating granuloma formation → potential fibrosis with chronicity | Th1-type inflammation; ↑ IL-2, interferon-γ; macrophage activation; TNF-α–mediated granuloma formation; defective antigen clearance | Pulmonary sarcoidosis or berylliosis |
| Alveolar-filling ILD [4] | Induced epithelial injury → pigmented macrophage accumulation → airway-centered inflammation ± fibrosis | Macrophage-driven inflammation, leading to alveolar and small airway filling defects | Cryptogenic organizing pneumonia, respiratory bronchiolitis ILD, or alveolar macrophage pneumonia (previously known as desquamative interstitial pneumonia)[34] |
| Idiopathic nonspecific interstitial pneumonia (NSIP)[35] | Homogeneous interstitial inflammation ± fibrosis without UIP features | Lymphocytic alveolitis; fibroblast foci less prominent than UIP; immune-mediated | Fibrotic and nonfibrotic NSIP |
| Lymphoid interstitial pneumonia (LIP)[4] | Polyclonal lymphocytic infiltration of the interstitium → cystic changes | Autoimmune dysregulation; CD4+ lymphocyte infiltration; association with Sjögren disease | Idiopathic or secondary LIP |
| Diffuse alveolar damage (DAD) or previously acute interstitial pneumonia (AIP)[36] | DAD often postinfection or trigger → acute lung injury | Endothelial & epithelial injury; hyaline membrane formation; cytokine storm | DAD, AIP (Hamman–Rich syndrome) |
|
Progressive pulmonary fibrosis, non-IPF fibrotic ILD |
Shared fibrotic pathway irrespective of original etiology | TGF-β-driven fibroblast activation, ECM accumulation, progressive architectural destruction despite treatment | Fibrotic HP, fibrotic NSIP, RA-ILD, SSc-ILD, unclassifiable ILD when meeting PPF criteria |
Histopathology
Histopathological examination plays a central role in diagnosing ILD, particularly when high-resolution computed tomography findings are nondiagnostic or do not align with a specific pattern. Surgical lung biopsy (via video-assisted thoracoscopic surgery) remains the gold standard for tissue diagnosis, though transbronchial lung cryobiopsy is an emerging minimally invasive alternative that is increasingly used in experienced ILD centers.[23]
Usual Interstitial Pneumonia
Usual interstitial pneumonia (UIP) is the histological pattern of IPF and is the most important pattern to recognize (see Image. Pattern of Usual Interstitial Pneumonia/Idiopathic Pulmonary Fibrosis on Computed Tomography Scan). Key features include temporal and spatial heterogeneity (areas of normal lung adjacent to honeycombing and fibroblastic foci), subpleural and basal predominance, fibroblastic foci (active sites of fibrogenesis at the leading edge of fibrosis), honeycombing with or without peripheral traction bronchiectasis, and relative absence of significant granulomas or extensive ground-glass opacity (see Image. Histopathology of Usual Interstitial Pneumonia in Idiopathic Pulmonary Fibrosis). Hyaline membranes and diffuse alveolar damage are absent in UIP, distinguishing UIP from acute exacerbation histology (see Image. Computed Tomography of the Lungs in Usual Interstitial Pneumonia).[37]
Nonspecific Interstitial Pneumonia
NSIP is characterized by temporal homogeneity (all areas of the lung appear similar in age), bilateral symmetric interstitial inflammation and/or fibrosis, and relative subpleural sparing, in contrast to UIP. Two subtypes exist: cellular NSIP (predominantly lymphoplasmacytic infiltration, better prognosis) and fibrotic NSIP (collagen deposition, worse prognosis but better than UIP). NSIP is the most common pattern in CTD-ILD, particularly SSc-ILD and antisynthetase syndrome (see Image. Nonspecific Interstitial Pneumonia).[38]
Organizing Pneumonia
Organizing pneumonia demonstrates intraluminal granulation tissue (Masson bodies) within alveolar ducts and alveoli, while preserving the underlying lung architecture. The pattern is temporally homogeneous and patchy. Importantly, organizing pneumonia is characterized by its reversibility with corticosteroid treatment, a key distinguishing feature. Cryptogenic organizing pneumonia is the idiopathic form; secondary organizing pneumonia may be drug-induced, infection-related, or associated with CTD (see Image. Organizing Pneumonia).[39]
Diffuse Alveolar Damage
Diffuse alveolar damage is the histological correlate of acute respiratory distress syndrome and acute interstitial pneumonia, also known as Hamman-Rich syndrome. The exudative phase is characterized by hyaline membrane formation, edema, and inflammatory infiltrates. The organizing and proliferative phase shows type II pneumocyte hyperplasia, interstitial fibrosis, and squamous metaplasia. Diffuse alveolar damage in the context of established ILD (particularly IPF) defines an acute exacerbation, which carries a very poor prognosis with in-hospital mortality exceeding 50%.[40]
Hypersensitivity Pneumonitis
Acute hypersensitivity pneumonitis shows bronchiolocentric lymphocytic infiltration with poorly formed granulomas and giant cells. Chronic fibrotic hypersensitivity pneumonitis demonstrates upper-lobe-predominant peribronchovascular fibrosis with bridging fibrosis, lymphocytic infiltration, and scattered poorly formed granulomas or giant cells. The presence of granulomas or giant cells adjacent to areas of fibrosis is the key feature distinguishing fibrotic hypersensitivity pneumonitis from UIP and IPF. This distinction is clinically important because it suggests a potentially treatable etiology through antigen identification and avoidance (see Image. Hypersensitivity Pneumonitis).
Desquamative Interstitial Pneumonia, Respiratory Bronchiolitis-ILD, and Alveolar Macrophage Pneumonia
These smoking-related ILDs share intraalveolar macrophage accumulation as their defining feature. Desquamative interstitial pneumonia (now relabeled alveolar macrophage pneumonia) shows diffuse, uniform accumulation of pigmented macrophages throughout alveolar spaces. Respiratory bronchiolitis-ILD shows bronchiolocentric macrophage accumulation in a more patchy distribution. Both are strongly associated with smoking and may stabilize or improve with smoking cessation alone.[41]
Lymphoid Interstitial Pneumonia
Lymphoid interstitial pneumonia (LIP) demonstrates dense polyclonal lymphocytic infiltration of the interstitium, with germinal center formation and cystic changes. LIP is associated with Sjögren disease, human immunodeficiency virus infection, and other autoimmune conditions. Distinction from lymphoma (monoclonal B-cell proliferation) requires immunohistochemistry and clonality studies (see Image. Lymphoid Interstitial Pneumonia).
Role of Transbronchial Lung Cryobiopsy
Transbronchial lung cryobiopsy (TBLC) uses a cryoprobe to obtain larger, better-preserved biopsy samples compared to conventional forceps transbronchial biopsy, with a reported diagnostic yield of 70% to 80% in ILD, approaching that of surgical lung biopsy in experienced centers. American Thoracic Society and European Respiratory Society 2022 guidelines conditionally recommend TBLC as an alternative to surgical biopsy in patients with nondiagnostic high-resolution computed tomography findings in experienced ILD centers. Complications include pneumothorax (approximately 5% to 10%) and bleeding; the surgical procedure should be performed with fluoroscopic guidance and bronchial blocker occlusion.[42]
Toxicokinetics
Toxicokinetics in interstitial lung disease primarily relates to drug-induced ILD and inhaled toxin-mediated pulmonary injury. More than 350 pharmaceutical agents have been identified as potential causes of ILD, and the mechanisms by which they cause pulmonary toxicity are diverse (see Image. Drug-Induced Interstitial Lung Disease).[43][44][45]
Mechanisms of Drug-Induced ILD
- Direct cytotoxicity: Reactive oxygen species generation damages alveolar epithelial cells and endothelium. This is the primary mechanism for drugs such as bleomycin, amiodarone, and nitrofurantoin. Bleomycin accumulates in the lung due to low concentrations of bleomycin hydrolase (the inactivating enzyme) in pulmonary tissue, leading to iron-catalyzed generation of free radicals and DNA strand breaks.
- Immune-mediated hypersensitivity: T-cell–mediated (type IV) hypersensitivity reactions cause lymphocytic alveolitis and organizing pneumonia patterns. Methotrexate, nitrofurantoin, and checkpoint inhibitor immunotherapy (programmed death-1 and programmed death-ligand 1 [PD-1 and PD-L1] inhibitors) cause ILD predominantly through this mechanism. Immune checkpoint inhibitor-related pneumonitis has emerged as a clinically important drug-induced ILD subtype, occurring in 3% to 5% of patients receiving anti-PD-1 therapy.
- Phospholipidosis: Amiodarone accumulates in lysosomes of type II pneumocytes and alveolar macrophages, inhibiting phospholipase enzymes and causing accumulation of phospholipids. This mechanism is responsible for the characteristic foamy macrophage appearance on lung biopsy in amiodarone pulmonary toxicity. Amiodarone's very long half-life (40 to 55 days) means pulmonary toxicity may persist or progress even after drug discontinuation.
- Vascular toxicity: Some chemotherapeutic agents (cyclophosphamide, carmustine) and targeted therapies cause endothelial injury and vascular remodeling, leading to pulmonary hypertension and ILD.[10]
Inhaled Toxin Mechanisms
Occupational and environmental inhalation of mineral dusts and organic antigens leads to ILD through distinct toxicokinetic pathways:
- Silica: Crystalline silica particles (quartz, cristobalite) are incompletely cleared by alveolar macrophages, leading to inflammasome (NLRP3) activation, interleukin-1β (IL-1β) and TNF-α release, and progressive granulomatous and fibrotic reaction. Silica's durability in biological tissue underpins the progressive nature of silicosis even after exposure cessation.[46]
- Asbestos: Asbestos fibers (particularly long, thin amphibole fibers) resist phagocytic clearance, stimulate reactive oxygen species and inflammatory cytokines, and form asbestos bodies (iron-coated fibers) that are pathological markers of significant exposure. Long fibers penetrate to the pleura, causing mesothelioma in addition to asbestosis.
- Organic antigens: Inhaled organic antigens (avian proteins, fungal spores) are processed by antigen-presenting cells and activate both immune complex (type III) and T-cell–mediated (type IV) pathways. The dose, frequency, and duration of antigen exposure, as well as individual immune susceptibility, determine whether acute, subacute, or chronic hypersensitivity pneumonitis develops.
Clinically Important Drug-ILD Associations
Key drug-ILD associations to recognize are as follows:
- Amiodarone: Prevalence of pulmonary toxicity 5% to 10% with long-term use; insidious onset; bilateral opacities on CT; foamy macrophages on bronchoalveolar lavage; may progress despite discontinuation
- Methotrexate: Hypersensitivity pneumonitis pattern; occurs at any dose; rechallenge generally contraindicated
- Nitrofurantoin: Acute (within weeks) or chronic (>6 months) patterns; one of the most common drug-induced ILD causes in primary care
- Checkpoint inhibitors (anti-PD-1/PD-L1): Pneumonitis in 3% to 5% of patients; treated with corticosteroids and immune checkpoint inhibitor discontinuation; may be severe (grade 3 to 4) in up to 1% to 2% of cases [47]
- Bleomycin: Cumulative dose-dependent (highest risk >400 units); supplemental oxygen use and renal impairment increase risk
A comprehensive drug history, including over-the-counter and herbal medications, is essential in any ILD evaluation. The pneumotox.com database provides a regularly updated reference of drug-lung toxicity associations and their evidence levels.
History and Physical
The initial and most crucial step in evaluating a patient suspected of having ILD is to gather a comprehensive history. Thoroughly documenting the patient's past medical history is vital during the initial assessment, because the cause of the disease is often identified through the patient's history. Besides a detailed review of previous systemic conditions and human immunodeficiency virus risk factors, several other historical aspects are particularly significant and include age, onset of symptoms, family history, and social, environmental, and occupational history (such as exposure to fumes, dust, or toxic inhalation), in addition to medication and past medical history, such as radiation.[48]
Symptoms are often nonspecific, with the most frequently noted being a slow development of breathlessness, particularly in those with scleroderma ILD and chronic hypersensitivity pneumonitis. Approximately 30% of patients with ILD report a nonresolving cough as the initial symptom. Although pleuritic chest pain is rare, chest pain can be present in specific subtypes like sarcoidosis. Hemoptysis might result from diffuse alveolar hemorrhages. Conversely, a patient may be entirely symptom-free yet exhibit abnormal imaging findings. The patient's history should include information about potential environmental or occupational exposures, current and past medications, and any exposure to radiation, fumes, dust, or toxic inhalants.
A family history is crucial because genetics might play a part. Symptoms of rheumatologic diseases should be taken into account; however, clinicians should recognize that shortness of breath may be the sole symptom of rheumatological-associated ILD.[14] Certain ILD types are more prevalent in specific age groups or are predominant in one sex. For example, most patients with sarcoidosis, connective tissue disease-associated ILD, lymphangioleiomyomatosis, and pulmonary Langerhans cell histiocytosis are typically between the ages of 20 and 40 years. In contrast, most patients with IPF are older than 50 years.
On physical examination, bibasilar fine end-inspiratory crackles (described as Velcro-like crepitations) are characteristic but not consistently observed. Patients with advanced disease may exhibit physical signs of pulmonary hypertension, such as an increased intensity of P2 of the second heart sound, elevated jugular venous pressure, and peripheral edema.[30] In patients with fibrotic ILD, digital clubbing is as common as nearly half of the cases, and could be a marker of poor prognosis (see Image. Clinical Examination Findings of Interstitial Lung Disease).[31]
Hypoxia at rest, cyanosis, and weight loss are common in advanced cases and may indicate a poor prognosis. In patients with suspected CTD-ILD, signs of active inflammatory arthritis, skin thickening, mechanic hands (cracked, hyperpigmented skin over lateral and dorsal digits), or Gottron papules (violaceous papules over dorsal metacarpophalangeal and interphalangeal joints) should be sought. In patients with suspected sarcoidosis, examination should include the skin for erythema nodosum and lupus pernio (violaceous plaques on the nose and cheeks) (see Image. Lupus Pernio), and the eyes for uveitis (see Image. Uveitis Associated with Interstitial Lung Disease).
Evaluation
The diagnosis of ILD can be established with appropriate history and exam, clinical findings, and, in most instances, should be confirmed after excluding other potential causes. ILD encompasses a broad spectrum of disorders, each requiring distinct management strategies and presenting varying prognoses, underscoring the importance of an accurate diagnosis. The diagnostic process commences with a comprehensive patient history and physical examination, supplemented by laboratory tests, imaging, physiological assessments, and potentially a biopsy (see Image. Diagnosis of Interstitial Lung Disease).[49] The initial routine laboratory evaluation includes a complete blood count to identify indicators such as hemolytic anemia, which may be associated with systemic lupus erythematosus, or eosinophilia, which could indicate drug-induced conditions.
A comprehensive CTD-ILD serology panel should include antinuclear antibody with reflex testing, rheumatoid factor, anti-cyclic citrullinated peptide, anti-Scl-70, anti-centromere, anti-RNA polymerase III (for SSc), myositis panel including anti-Jo-1, anti-melanoma differentiation-associated protein 5 (anti-MDA-5; associated with rapidly progressive ILD), and antisynthetase antibodies (anti-PL-7, PL-12, EJ, OJ), anti-Ro and anti-La (Sjögren disease), and antineutrophil cytoplasmic antibody (vasculitis-associated ILD). Anti-MDA5 antibody is of particular clinical urgency because it is associated with a rapidly progressive and often fatal ILD phenotype in dermatomyositis. In certain instances, evaluations for infectious diseases, such as HIV or hepatitis, may be warranted.[50]
The imaging workup begins with a standard chest radiograph, where the most common radiographic feature is a reticular pattern, although nodular or mixed patterns may also be observed (see Image. Diffuse Interstitial Lung Disease).[51] However, the sensitivity of a chest radiograph is lower than that of high-resolution computed tomography (HRCT), at approximately 60%.[52] In certain instances, specific patterns can narrow diagnostic possibilities. The presence of mediastinal lymphadenopathy on a chest radiograph may suggest lymphoma or sarcoidosis (see Image. Sarcoidosis Imaging). HRCT provides a more detailed characterization of the disease and can assist in diagnosis when chest radiograph results are negative; HRCT should be performed in the supine and prone positions.
HRCT should be performed in the supine and prone positions. In some cases, HRCT may reveal a classic radiological pattern of a disease such as UIP (see Images. Usual Interstitial Pneumonia on Axial High-Resolution Computed Tomography and Usual Interstitial Pneumonia). The typical radiological pattern of UIP on HRCT includes subpleural and basilar predominant changes, reticular patterns, and honeycomb changes, with or without traction bronchiectasis. Table 3 summarizes the radiologic changes in common types of ILD (see Image. High-Resolution Computed Tomography Patterns of Interstitial Lung Disease).
Table 3. Radiologic Changes in Common Types of Interstitial Lung Disease
| ILD Type | Radiologic Findings | Distribution | Additional Features |
| Idiopathic pulmonary fibrosis (usual interstitial pneumonia pattern) | Reticulation, honeycombing, and traction bronchiectasis | Basal and subpleural | Absence of significant ground-glass opacities; architectural distortion |
| Nonspecific interstitial pneumonia | Ground-glass opacities, fine reticulation | Bilateral, symmetric, lower lung zones | Relative subpleural sparing; minimal honeycombing |
| Alveolar macrophage pneumonia | Diffuse ground-glass opacities | Lower lung zones, peripheral | Smoking-related; limited fibrosis |
| Respiratory bronchiolitis–associated ILD | Centrilobular nodules, ground-glass opacities | Upper lobes | Strong association with smoking; mild disease |
| Hypersensitivity pneumonitis (chronic) | Ground-glass opacities, mosaic attenuation, fibrosis | Upper and mid lung zones | Air trapping on expiratory imaging |
| Sarcoidosis | Perilymphatic nodules, hilar lymphadenopathy | Upper and mid lung zones | Nodules along bronchovascular bundles |
| Pulmonary Langerhans cell histiocytosis | Cysts and nodules | Upper lung zones | Sparing of costophrenic angles; smoking-related |
| Organizing pneumonia | Patchy consolidations, ground-glass opacities | Peripheral and peribronchial | Reversible with treatment; migratory infiltrates |
| Lymphoid interstitial pneumonia | Ground-glass opacities, thin-walled cysts | Lower lung zones | Associated with autoimmune disease and HIV |
| Asbestosis | Reticulation, subpleural fibrosis | Lower lung zones | Pleural plaques are characteristic [4][24][28][33][34][35][38][53][54][55][56][57] |
Interdisciplinary discussion, involving pulmonologists, radiologists, and pathologists, is the diagnostic gold standard for ILD and is particularly essential when HRCT and histological findings are discordant or nondiagnostic. The American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Association (ATS/ERS/JRS/ALAT) guidelines recommend interdisciplinary discussion in all cases where the diagnosis remains uncertain after a comprehensive noninvasive workup.[23] Table 4 describes the monitoring required.
If the diagnosis remains uncertain after evaluating the patient's history, laboratory results, and radiological findings, an invasive workup may be advantageous. Bronchoalveolar lavage (BAL) yields nonspecific results because no findings in a BAL are definitively pathognomonic for a specific type of ILD. However, BAL can be instrumental in narrowing down the options. For example, in patients thought to have hypersensitivity pneumonitis, BAL will demonstrate marked lymphocytosis. The decision to pursue a lung biopsy should be individualized, because not all cases necessitate one. Lung biopsy is most beneficial in diagnosing sarcoidosis and idiopathic interstitial pneumonia. Guidelines from the ATS suggest that a surgical lung biopsy should be considered when a diagnosis of IPF cannot be established noninvasively, particularly if clinical and radiological evidence does not align with UIP, unless the patient is at high risk of complications.[22]
TBLC is conditionally recommended by ATS/ERS 2022 guidelines as a less invasive alternative to surgical lung biopsy in experienced ILD centers, with a diagnostic yield of approximately 70% to 80% and a lower procedural risk profile than video-assisted thoracoscopic surgery. Complete pulmonary function tests (PFT), including assessment of forced vital capacity, diffusion capacity, and oxygenation, are important for assessing disease severity. Individuals with ILD often present with a restrictive ventilatory pattern on spirometry. Oximetry is essential for all patients with ILD for prognostication and disease monitoring. However, a complete functional assessment should include the 6-minute walk test (6MWT).[58] Results from an observational study found that the 6MWT correlates closely with PFTs in patients with ILD. These functional tests also help in monitoring the progression of ILD, not only in establishing the diagnosis.
When to Screen
- Perform screening in patients with systemic associated rheumatological disorders who have respiratory signs or symptoms suggestive of ILD
- Consider screening in asymptomatic patients with high-risk features listed above
Initial Screening Tests
- PFTs: spirometry, lung volumes, and diffusion capacity
If ILD is suspected or the risk is increased
• Repeat PFT• Ambulatory oxygen desaturation testing• HRCT of the chest as clinically indicated
Table 4. Monitoring After Initial Screening
| Disease group | Recommended monitoring frequency |
| Idiopathic inflammatory myopathies | PFTs every 3–6 months during the first year, then less frequently once stable |
| Systemic sclerosis | PFTs every 3–6 months during the first year, then less frequently once stable |
| Rheumatoid arthritis, Sjögren disease, MCTD | PFTs every 3–12 months during the first year, then less frequently once stable [59] |
MCTD, mixed connective tissue disease; PFT, pulmonary function test
Other Tests
- Ambulatory desaturation testing: Every 3 to 12 months
- HRCT chest: As needed based on symptoms, PFT decline, or clinical concern
Treatment / Management
Pharmacological Therapy
Pharmacological therapy depends on the ILD class and on eliminating the specific cause. In most idiopathic cases, therapy is supportive. However, the mainstay therapy for the treatment of IPF is novel antifibrotic therapies.[60] Nintedanib and pirfenidone are tyrosine kinase inhibitors approved for the treatment of IPF.[61] Research results indicate that both drugs can slow disease progression. With progressive disease despite the elimination of the offending agent, corticosteroids are indicated. Patients with cryptogenic organizing pneumonia or hypersensitivity pneumonitis can demonstrate a rapid, dramatic improvement with corticosteroids. For patients who do not respond to corticosteroids, immunosuppressant therapy is commonly considered. Table 5 summarizes treatment options for ILD by subtype.
Table 5. Treatment Options for Interstitial Lung Disease by Subtype
| ILD Subtype | First-Line Treatment | Alternative or Adjunct Treatment | Key Notes |
| IPF | Nintedanib or pirfenidone | Lung transplant for advanced disease; inhaled treprostinil for associated pulmonary hypertension | Antifibrotic therapy reduces annual FVC decline by approximately 44% to 57% and is strongly recommended by ATS/ERS/JRS/ALAT guidelines |
| SSc-ILD | MMF | Tocilizumab, nintedanib, cyclophosphamide, rituximab; MMF and nintedanib combination; corticosteroids are not recommended | MMF and cyclophosphamide stabilize lung function over 52 weeks; tocilizumab reduces FVC decline; MMF is strongly recommended by ATS |
| CTD-ILD | Mycophenolate mofetil | Rituximab, tocilizumab, cyclophosphamide, azathioprine; nintedanib for progressive disease | Immunomodulatory therapy may stabilize or improve FVC over 12 months |
| PPF | Nintedanib (strong recommendation); pirfenidone (conditional) | Immunomodulatory therapy when inflammatory features are present | Results from the INBUILD trial showed nintedanib slows FVC decline in PPF of diverse underlying etiology at 52 weeks—the first antifibrotic with demonstrated benefit across multiple non-IPF fibrotic ILD subtypes |
| Hypersensitivity pneumonitis | Antigen avoidance; corticosteroids | Mycophenolate mofetil, rituximab, azathioprine, and nintedanib for progressive fibrotic disease | Immunosuppression benefits inflammatory disease; antifibrotics are indicated for progressive fibrotic phenotype |
| Sarcoidosis | Prednisone | Methotrexate or azathioprine (second-line); anti-TNF therapy or JAK inhibitors for refractory disease | Corticosteroids: recommended for pulmonary disease with a treatment indication |
| RA-ILD | Immunomodulatory therapy (MMF, rituximab) | Nintedanib or pirfenidone for progressive fibrotic disease; tocilizumab in select cases | Limited RCT data; management extrapolated from CTD-ILD and PPF trials |
| ILD with pulmonary hypertension (WHO group 3) | Inhaled treprostinil | Optimization of ILD therapy by subtype | Inhaled treprostinil improves 6-minute walk distance and respiratory symptoms |
| End-stage ILD (any cause) | Lung transplant evaluation | Palliative care, oxygen therapy, and pulmonary rehabilitation | Median posttransplant survival ~5–7 years vs <2 years without transplant; early referral recommended [23][24][62][63][64][65][66] |
ALAT, Latin American Thoracic Association; ATS, American Thoracic Society; COP, cryptogenic organizing pneumonia; CTD-ILD, connective tissue disease–associated interstitial lung disease; ERS, European Respiratory Society; FVC, forced vital capacity; GERD, gastroesophageal reflux disease; HP, hypersensitivity pneumonitis; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; ISHLT, International Society for Heart and Lung Transplantation; JRS, Japanese Respiratory Society; MMF, mycophenolate mofetil; PPF, progressive pulmonary fibrosis; RA-ILD, rheumatoid arthritis–associated interstitial lung disease; RCT, randomized controlled trial; SSc-ILD, systemic sclerosis–associated interstitial lung disease; WHO, World Health Organization
ATS and International Society for Heart and Lung Transplantation guidelines recommend listing consideration when: FVC is less than 80% predicted and declining, diffusing capacity of the lung for carbon monoxide is less than 40% predicted, 6MWT distance is less than 250 m or declining, or there is an acute exacerbation of ILD. Early referral (at the time of IPF diagnosis) is recommended because waitlist times can be 2 to 4 years.[67] Newly developed drugs such as nerandomilast (BI 1015550) are orally administered and selectively inhibit phosphodiesterase 4B, providing antifibrotic and immunomodulatory effects.
In a phase 3 multicenter study of 1177 patients with IPF from more than 30 countries, participants were either receiving stable antifibrotic treatment (pirfenidone or nintedanib) or had been off treatment for 8 weeks.[68] Participants were 40 years of age or older, with an FVC of at least 45% and a diffusing capacity of the lung for carbon monoxide of at least 25% of predicted. Nerandomilast achieved its primary endpoint by showing a significantly smaller FVC reduction versus placebo, with only the 18-mg twice-daily dose proving effective. The drug showed no impact on the risk of acute exacerbation, respiratory-related hospitalization, or mortality. The main adverse effect was diarrhea, causing drug discontinuation in less than 4% of patients, and occurring more frequently when combined with nintedanib therapy.
Nonpharmacological Management Strategies for Interstitial Lung Disease
Supportive care
- Providing supplemental oxygen to patients with hypoxemia at rest or during exertion if the arterial oxygen saturation is less than 88%
- Implementing pulmonary rehabilitation to improve exercise capacity, reduce dyspnea, and enhance quality of life
Management of comorbidities
- Treating pulmonary hypertension with targeted therapies when appropriate [69]
- Managing gastroesophageal reflux disease through lifestyle changes and medications [70]
- Assessing and treating obstructive sleep apnea with proper sleep studies and positive airway pressure therapy, because up to two-thirds of patients with ILD have comorbid obstructive sleep apnea [71]
- Following lung cancer screening guidelines and participating in interdisciplinary cancer care (A1)
Symptom control and advanced care
- Offering palliative care for persistent symptoms like dyspnea, cough, fatigue, and emotional distress
- Integrating palliative services early to improve symptom management and facilitate end-of-life planning
Some studies' results have provided indirect evidence that early therapy during the course of the disease might correlate with therapeutic responsiveness, as the lung architecture has not suffered significant derangement. Once fibrosis initiates, there is no treatment to reverse it, but nintedanib can slow disease progression.[49] Transplantation is the only treatment that can restore physiologic function.
Results from some studies have provided indirect evidence that early therapy during the course of the disease might correlate with therapeutic responsiveness, as the lung architecture has not yet suffered significant derangement. Once fibrosis has initiated, no treatment has been shown to reverse it, but nintedanib can slow disease progression.[49] A lung transplant is the only treatment modality that can restore physiological function in patients.
Differential Diagnosis
The differential diagnosis of ILD encompasses both conditions that mimic ILD radiographically and the critical distinction between ILD subtypes, which has direct therapeutic implications.
Conditions That Mimic ILD
- Pulmonary edema (cardiogenic): Bilateral interlobular septal thickening, ground-glass opacity, and Kerley B lines on HRCT may simulate ILD. Distinguishing features include cardiomegaly on chest radiograph, pleural effusions, improvement with diuresis, elevated B-type natriuretic peptide and N-terminal pro-B-type natriuretic peptide levels, and bilateral perihilar (butterfly) distribution. Echocardiography is essential when a cardiac cause is suspected.
- Infectious pneumonias (bacterial, viral, fungal): Consolidation, ground-glass opacity, and nodules can overlap with ILD on imaging. Key distinguishing features include acute onset, fever, elevated inflammatory markers, and response to antimicrobial therapy. Pneumocystis jirovecii pneumonia in patients who are immunocompromised can simulate NSIP with diffuse bilateral ground-glass opacity. Bronchoalveolar lavage with silver stain or polymerase chain reaction is diagnostic.[72]
- Acute respiratory distress syndrome and diffuse alveolar damage: Acute onset bilateral opacification overlaps with acute ILD presentations. Acute respiratory distress syndrome is defined by a fraction of inspired oxygen (PaO2/FiO2) ratio less than 300, bilateral infiltrates, absence of cardiogenic cause, and onset within 7 days of a precipitating event.
- Lymphoma (pulmonary): Primary pulmonary lymphoma or lymphomatoid granulomatosis may present with consolidation, nodules, or ground-glass opacity that overlap with those of hypersensitivity pneumonitis, sarcoidosis, or organizing pneumonia. Tissue biopsy with clonality studies is definitive.
- Pulmonary alveolar proteinosis: The crazy-paving pattern on HRCT (ground-glass opacity with superimposed interlobular septal thickening) is characteristic and can mimic certain ILD patterns. Bronchoalveolar lavage with milky and opaque appearance and periodic acid-Schiff–positive material is diagnostic.[73]
Critical Intra-ILD Differential: Patterns With Therapeutic Implications
- UIP (IPF) versus fibrotic NSIP versus fibrotic hypersensitivity pneumonitis: This is the most clinically important differential for ILD. UIP and IPF are characterized by subpleural honeycombing, basal predominance, minimal ground-glass opacity, no upper lobe predominance, and no granulomas. Fibrotic NSIP demonstrates subpleural sparing, temporal homogeneity, more ground-glass opacity, and CTD association. Fibrotic HP shows predominance of the upper and mid-lobes, lobular air trapping, a history of antigen exposure, and poorly formed granulomas on biopsy. Misclassification leads to incorrect treatment: antifibrotics for IPF and PPF versus immunosuppression for fibrotic HP.
- Sarcoidosis versus hypersensitivity pneumonitis versus lymphoma: All may present with bilateral hilar lymphadenopathy and upper- and mid-lung nodules. Sarcoidosis demonstrates perilymphatic nodules, bilateral symmetric hilar adenopathy, elevated angiotensin-converting enzyme levels, and noncaseating granulomas on biopsy. HP shows centrilobular nodules, air trapping, a history of antigen exposure, and BAL lymphocytosis. Lymphoma presents with constitutional symptoms and monoclonal proliferation on biopsy.
- CTD-ILD versus IPF: CTD-ILD should be specifically excluded before diagnosing IPF, because management differs significantly. Clinicians should perform a thorough CTD evaluation in younger women with NSIP pattern, autoimmune symptoms (dry eyes, Raynaud phenomenon, arthritis, skin changes), or elevated autoantibody levels. Interstitial pneumonia with autoimmune features is a research category for patients with autoimmune features not meeting full CTD diagnostic criteria.[74]
- Acute exacerbation of IPF versus infection versus pulmonary edema: New bilateral ground-glass opacity superimposed on an established fibrotic pattern in patients with IPF should prompt urgent workup to exclude infection and cardiac cause before diagnosing acute exacerbation of IPF. An acute exacerbation carries a greater than 50% in-hospital mortality and requires high-dose corticosteroids, but immunosuppression in the context of pneumonia can be fatal.
Pertinent Studies and Ongoing Trials
Landmark Completed Trials
- CAPACITY and ASCEND trials (pirfenidone, IPF): Results from 2 phase 3 trials (Clinical Assessment of Three Doses of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis [CAPACITY]; Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis [ASCEND]) established pirfenidone as the first antifibrotic approved for IPF. Pirfenidone reduced annual FVC decline by 47.9% compared to placebo in ASCEND (mean difference 235 mL/year). Both trials demonstrated reduced disease progression without a survival benefit at the individual trial level.[23][75]
- INPULSIS trials (nintedanib, IPF): Results from INPULSIS-1 and INPULSIS-2 demonstrated that nintedanib reduced the annual decline in FVC by approximately 125 to 130 mL/year compared with placebo. INPULSIS-2 also showed reduced exacerbation risk. Nintedanib and pirfenidone are both strongly recommended by ATS, ERS, JRS, and ALAT guidelines as equivalent first-line antifibrotic options in IPF.[76]
- SENSCIS trial (nintedanib, SSc-ILD): Results from this phase 3 randomized controlled trial of 576 patients with SSc-ILD (Safety and Efficacy of Nintednib in Systemic Sclerosis [SENCIS] showed nintedanib reduced annual FVC decline by 44% versus placebo (-52.4 versus -93.3 mL/year; P < .04). This trial established nintedanib as the first pharmacological therapy with demonstrated benefit in SSc-ILD, leading to the United States Food and Drug Administration approval in 2019.[63]
- INBUILD trial (nintedanib, PPF): Results from this phase 3 RCT of 663 patients with progressive pulmonary fibrosis of diverse underlying etiologies (excluding IPF) showed that nintedanib reduced the annual decline in FVC compared with placebo (-80.8 versus -187.8 mL/year; P < .001), regardless of HRCT fibrosis pattern. This landmark trial established nintedanib as the first antifibrotic approved for PPF across multiple non-IPF fibrotic ILD subtypes, representing a major paradigm shift in the treatment of progressive fibrosis.[77]
- SLS II trial (mycophenolate vs cyclophosphamide, SSc-ILD): Results from this phase 3 randomized controlled trial of 142 patients with SSc-ILD showed that mycophenolate mofetil was noninferior to cyclophosphamide in improving FVC at 24 months, with a more favorable toxicity profile. This trial established mycophenolate mofetil as the preferred first-line immunosuppressive agent for SSc-ILD, replacing cyclophosphamide.[78]
- Idiopathic Pulmonary Fibrosis International Group Exploring N-Acetylcysteine (IFIGENIA) trial (N-acetylcysteine + prednisone + azathioprine vs prednisone + azathioprine, IPF): Demedts et al established the use of N-acetylcysteine in IPF. Subsequently, in the Prednisone, Azathioprine, and N-Acetylcysteine: A Study That Evaluates Response in Idiopathic Pulmonary Fibrosis (PANTHER-IPF) trial, Raghu et al discontinued triple therapy (prednisone + azathioprine + N-acetylcysteine) early due to increased mortality, fundamentally changing IPF management away from immunosuppression toward antifibrotic therapy.[79][80]
- FIBRONEER-IPF trial (nerandomilast [BI 1015550], IPF): Results from this phase 3 RCT of 1177 patients with IPF showed that nerandomilast (a phosphodiesterase 4B [PDE4B] inhibitor) demonstrated a significantly smaller FVC reduction than placebo at 52 weeks, with an acceptable safety profile. As of early 2026, nerandomilast is under regulatory review.[68][81]
Key Ongoing Trials
- BMS-986325 (TGF-β inhibitor, IPF): This phase 2 trial targets the central profibrotic cytokine TGF-β, which drives fibroblast activation and ECM accumulation across multiple ILD subtypes.
- ISABELA 1&2 trials (ziritaxestat, IPF): These phase 3 trials of an autotaxin inhibitor were discontinued due to an unfavorable benefit-to-risk profile, demonstrating the challenges of targeting the LPA pathway in IPF.
- Phase 3 trials of inhaled treprostinil (INCREASE trial expansion): Building on the INCREASE trial, inhaled treprostinil improved 6MWT in ILD-associated pulmonary hypertension. Trials are ongoing to define durability and optimal patient selection.
- Anti–connective tissue growth factor pamrevlumab: Phase 3 (ZEPHYRUS-1 and ZEPHYRUS-2) trials of pamrevlumab (anti-CTGF monoclonal antibody) in IPF, results pending.
Prognosis
Prognosis varies substantially across ILD subgroups and is determined by the underlying ILD subtype, histological pattern, functional parameters at diagnosis, the rate of physiological decline, and the presence of complications.
General Adverse Prognostic Indicators
Across all ILD subtypes, the following factors are associated with worse outcomes:
- Physiological parameters: Baseline FVC less than 70% to 75% of predicted; DLCO <40% predicted; resting hypoxia (SpO2 <88%); 6MWT distance <250 m or >50m decline over 6 months; FVC decline >10% or DLCO decline >15% in the preceding year [82]
- Radiological: Extent of fibrosis on CT >20%; presence of honeycombing; UIP pattern (versus NSIP or organizing pneumonia)
- Demographic: Age >60 years; male sex; higher body mass index
- Clinical events: Acute exacerbations of ILD requiring hospitalization are associated with markedly reduced survival; a median posthospitalization survival of approximately 7 months in acute exacerbation of IPF
Subtype-Specific Prognosis
- IPF: Median survival 2 to 5 years from diagnosis without treatment. The introduction of antifibrotic therapy (nintedanib, pirfenidone) slows FVC decline but does not alter the underlying progressive natural history for most patients. Five-year survival is approximately 20% to 40%. Lung transplant offers a median posttransplant survival of 5 to 7 years and is the only treatment that alters the natural history.[83]
- SSc-ILD: The most clinically severe CTD-ILD subtype. Median survival from SSc diagnosis is 11 years, but ILD is the leading cause of SSc-related mortality. Fibrotic pattern (UIP on HRCT), male sex, anti-Scl-70 positivity, and diffuse cutaneous SSc subtype are associated with more severe ILD and worse prognosis.
- CTD-ILD (rheumatoid arthritis, polymyositis/dermatomyositis, Sjögren disease, mixed connective tissue disease): Generally, a better prognosis than IPF. The NSIP pattern (common in CTD-ILD) has a better prognosis than UIP. Antimelanoma differentiation–associated gene 5-positive dermatomyositis-ILD is a notable exception because it is a rapidly progressive ILD phenotype with high short-term mortality if untreated.
- Hypersensitivity pneumonitis: Prognosis depends critically on identification and avoidance of the causative antigen. Acute and subacute hypersensitivity pneumonitis with an identifiable antigen that is successfully eliminated carries an excellent prognosis with near-complete resolution. Chronic fibrotic HP with UIP pattern carries a prognosis similar to IPF.
- Sarcoidosis: Overall, good prognosis, and 60% of patients achieve spontaneous remission within 2 to 3 years. Approximately 20% develop chronic disease, and 5% progress to severe pulmonary fibrosis. Löfgren syndrome (acute sarcoidosis with erythema nodosum, bilateral hilar adenopathy, arthritis) has an excellent prognosis with near-universal spontaneous resolution.
- Organizing pneumonia (cryptogenic organizing pneumonia): Excellent response to corticosteroids (80% to 90% of patients respond). Relapses occur in approximately 50% of patients when corticosteroids are tapered too rapidly, but usually respond to retreatment. Progression to irreversible fibrosis is very rare.
ILD-Associated Pulmonary Hypertension as a Prognostic Modifier
Pulmonary hypertension complicates up to 30% to 40% of patients with advanced ILD and is an independent predictor of mortality. The development of ILD-associated pulmonary hypertension (World Health Organization Group 3) markedly worsens prognosis. The median survival is approximately 1 year from diagnosis in IPF-associated pulmonary hypertension without treatment. Inhaled treprostinil (INCREASE trial) improves 6MWT distance and reduces clinical worsening, and represents the first approved therapy for this complication
Complications
ILS is associated with a range of serious complications that contribute significantly to morbidity and mortality. Recognition and proactive treatment of these complications are essential to optimizing patient outcomes.
Progressive Respiratory Failure and Hypoxia
Progressive dyspnea and hypoxia are universal complications of advancing ILD. Resting hypoxia (oxygen saturation, SpO2 < 88%) develops as fibrosis replaces functional parenchyma, leading to ventilation-perfusion mismatch and impaired gas transfer (DLCO decline). Supplemental oxygen is recommended when SpO2 decreases below 88% at rest or with exertion, improving exercise tolerance, dyspnea, and quality of life, though the effect on survival in ILD has not been definitively established.
Acute Exacerbation of ILD
Acute exacerbation (AE)-ILD is defined as a new bilateral ground-glass opacity superimposed on a preexisting fibrotic pattern with no identifiable cause (excluding infection, cardiac failure, or volume overload) and is the most feared complication of IPF and other fibrotic ILDs. AE-IPF carries an in-hospital mortality exceeding 50%, with a median survival of less than 3 months from the event. High-dose corticosteroids (methylprednisolone 500 mg to 1000 mg/d for 3 days) are commonly prescribed, though evidence for this practice is limited. Mechanical ventilation provides only temporary stabilization; most patients do not recover to baseline function.
Pulmonary Hypertension (ILD-PH, WHO Group 3)
Pulmonary hypertension develops in 30% to 40% of patients with advanced interstitial lung disease due to hypoxic vasoconstriction, vascular remodeling, and fibrosis-induced compression of the pulmonary vasculature. ILD-PH is an independent predictor of mortality, with a median survival of approximately 1 year after diagnosis in IPF-PH. Clinical recognition, as suggested by progressive dyspnea disproportionate to lung function decline, syncope, and right heart strain on electrocardiography, should prompt echocardiography and right heart catheterization. Inhaled treprostinil (INCREASE trial) is the only United States Food and Drug Administration–approved therapy for ILD-PH, and results from the INCREASE trial demonstrated improvement in 6-minute walk test distance and reduction in clinical worsening.
Lung Cancer
Patients with IPF have a 5% to 10% prevalence of concurrent lung cancer, substantially higher than in the general population with a smoking history. The shared risk factors (smoking, age) and the fibrotic lung environment (which promotes epithelial-mesenchymal transition and DNA damage from oxidative stress) account for this association. Lung cancers in patients with IPF most commonly arise in the periphery and lower lobes, areas of maximal fibrosis, and are predominantly squamous cell carcinoma or adenocarcinoma. Treatment is complicated by the elevated surgical risk (AE-ILD triggered by resection) and the potential for worsening ILD with platinum-based chemotherapy. Annual low-dose CT should be performed in all patients with a smoking history and ILD for lung cancer screening.
Infections
Patients with ILD are at increased risk of respiratory infections due to impaired mucociliary clearance, structural lung abnormalities (particularly honeycombing), and immunosuppressive therapy. Key infection risks include the following:
- Bacterial pneumonia: Higher incidence in patients with ILD; bacterial pneumonia may precipitate AE-ILD and carries high mortality. Annual influenza and pneumococcal vaccination are recommended for all patients with ILD.
- Opportunistic infections: Patients receiving immunosuppressive therapy (corticosteroids, azathioprine, cyclophosphamide, rituximab) are at risk for Pneumocystis jirovecii pneumonia and fungal infections. Prophylaxis (trimethoprim-sulfamethoxazole) should be considered in patients receiving 20 mg/d or more of prednisolone equivalent for 4 weeks or more in combination with another immunosuppressant.
Venous Thromboembolism
Patients with ILD, particularly those with IPF, have an elevated risk of venous thromboembolism (deep vein thrombosis and pulmonary embolism), likely driven by a chronic procoagulant state associated with progressive fibrosis and systemic inflammation. Pulmonary embolism can be clinically difficult to distinguish from AE-ILD and should be actively excluded in patients with acute deterioration. Appropriate prophylaxis during hospitalizations is essential.[84]
Gastroesophageal Reflux Disease and Aspiration
Gastroesophageal reflux disease is present in up to 90% of patients with IPF and may contribute to ILD pathogenesis and progression through chronic microaspiration of gastric acid contents. Aggressive treatment (proton pump inhibitors, head elevation, dietary modification) is recommended as part of IPF treatment. Antireflux surgery (fundoplication) may be considered in patients with IPF and severe refractory gastroesophageal reflux disease, though evidence for a direct ILD benefit is limited.
Obstructive Sleep Apnea
Obstructive sleep apnea complicates up to two-thirds of patients with ILD, contributing to nocturnal hypoxia, poor sleep quality, and exacerbation of pulmonary hypertension. Screening for obstructive sleep apnea should be performed in all patients with ILD, particularly those with unexplained nocturnal desaturation, daytime somnolence, or coexistent obesity. Positive airway pressure therapy improves oxygenation and quality of life in patients with ILD and concurrent obstructive sleep apnea.
Psychological Complications
Anxiety and depression are highly prevalent in patients with ILD (up to 30% to 40%), driven by progressive disability, oxygen dependence, and poor prognosis. These conditions are frequently underdiagnosed and undertreated. Psychological assessment should be part of routine ILD follow-up, and referral to psychological or palliative care services is appropriate.[85]
Postoperative and Rehabilitation Care
Pulmonary Rehabilitation
Pulmonary rehabilitation is a core component of ILD treatment and should be offered to all symptomatic patients (Medical Research Council dyspnea scale score >2), regardless of disease subtype. Evidence supports that pulmonary rehabilitation improves exercise capacity, dyspnea, quality of life, and psychological well-being in patients with ILD, though effects on long-term disease progression have not been demonstrated.
- Program components: Supervised exercise training (aerobic training and strength training), education (disease, medications, oxygen use, energy conservation), nutritional assessment, psychological support, and advanced care planning
- Exercise training: Moderate-intensity walking or cycling for 20 to 30 minutes per session, 3 to 5 times per week, supplemented with resistance training for peripheral muscle deconditioning. Supplemental oxygen should be provided during exercise if required to maintain SpO2 ≥88%
- Maintenance: Benefits of pulmonary rehabilitation are not sustained without ongoing exercise maintenance. Patients should be enrolled in community maintenance exercise programs following completion of supervised rehabilitation [86]
- Referral timing: Pulmonary rehabilitation should be initiated early in the disease course, before functional decline is severe, because patients with very advanced disease may not tolerate the exercise program
Consultations
Optimal ILD treatment requires a comprehensive interdisciplinary team approach. The complexity of ILD diagnosis and treatment, encompassing respiratory physiology, radiology, pathology, rheumatology, and increasingly oncology, necessitates coordinated specialist input.[87]
Core ILD Team: Required for All Patients
- Pulmonologist or interventional pulmonologist: Leads the ILD interdisciplinary team. Coordinates diagnostic workup, including HRCT, PFT, bronchoalveolar lavage (BAL), and tissue sampling (transbronchial lung cryobiopsy [TLBC] or surgical biopsy referral). Oversees antifibrotic and immunosuppressive treatment initiation and monitoring. Performs bronchoscopy and BAL for diagnostic workup and infection surveillance.
- Radiologist (thoracic): Expert thoracic radiology review of HRCT is essential because interobserver variability in ILD pattern recognition is high even among experienced radiologists. A dedicated thoracic radiologist experienced in ILD patterns is required for accurate classification of UIP, NSIP, HP, and sarcoidosis. HRCT was reviewed in the context of the clinical and pathological findings at the interdisciplinary discussion.
- Pathologist: Specialist lung pathology review of video-assisted thoracoscopic surgery, biopsy, or TBLC specimens for UIP, NSIP, OP, DAD, hypersensitivity pneumonitis, and other patterns. Immunohistochemistry for lymphocyte subsets, granuloma characterisation, and exclusion of infection or malignancy. Interdisciplinary input is essential when histological and radiological patterns are discordant.
Additional Specialist Consultations
- Rheumatologist: Essential for all patients with suspected CTD-ILD, including those with interstitial pneumonia with autoimmune features. Coordinates serological workup, CTD diagnosis, and treatment of systemic disease activity. Particularly important in SSc-ILD, RA-ILD, inflammatory myopathy–associated ILD, and antisynthetase syndrome.
- Cardiology and echocardiography: For assessment of pulmonary hypertension (echocardiogram to estimate right ventricular systolic pressure and right heart function), right heart catheterization when pulmonary hypertension is confirmed, and treatment of comorbid cardiac disease (atrial fibrillation, right heart failure) in advanced ILD.
- Transplant pulmonologist or thoracic surgeon: Referral for lung transplant evaluation should be made at the time of IPF diagnosis (not delayed until end-stage), given a median waitlist time of 2 to 4 years. All patients with progressive ILD receiving maximal medical therapy should be assessed for transplant eligibility. Surgical referral for video-assisted thoracoscopic surgery biopsy is appropriate when TBLC is unavailable or technically infeasible.
- Palliative care is recommended by ATS guidelines: Early integration of palliative care from the time of diagnosis in progressive ILD (particularly IPF). Palliative care provides specialist symptom treatment (refractory dyspnea, cough, anxiety), advanced care planning, goals of care discussions, and support for caregivers. Palliative care should not be reserved for end-of-life care.[88]
- Sleep medicine: For evaluation and treatment of obstructive sleep apnea, which complicates up to two-thirds of patients with ILD. Initiation of polysomnography and positive airway pressure therapy are appropriate referral indications.
- Gastroenterology: For evaluation and treatment of gastroesophageal reflux disease, which is present in up to 90% of patients with IPF and may contribute to ILD progression through microaspiration. Consideration of antireflux intervention in refractory cases is appropriate.
- Clinical pharmacist: For medication reconciliation (drug-ILD screening of all medications using pneumotox.com), monitoring of immunosuppressive drug adverse effects (cyclophosphamide bladder toxicity, mycophenolate gastrointestinal tract toxicity, azathioprine myelosuppression), dose adjustments for renal and hepatic impairment, and drug-drug interaction review.
- Specialist respiratory nurse or ILD nurse coordinator: The patient's primary point of contact. The ILD nurse coordinator coordinates clinic appointments, assists with inhaler technique, oxygen therapy education, provides patient and caregiver education on disease progression and advanced care planning, facilitates referrals, and provides psychological support.
- Dietitian: Nutritional assessment and support for patients with weight loss, muscle wasting, or inadequate intake, which is common in advanced ILD. Pulmonary cachexia is an adverse prognostic indicator. Adequate protein intake supports respiratory muscle function and exercise capacity during pulmonary rehabilitation.
- Psychology and psychiatry: For anxiety and depression (30% to 40% prevalence in ILD), adjustment disorder, and existential distress related to the diagnosis of a progressive life-limiting illness. Models of care from psycho-oncology apply to ILD.
Deterrence and Patient Education
Patient education is a fundamental component of ILD treatment, enabling patients to actively participate in their care, recognize warning signs, and make informed decisions about treatment and advance care planning.[23]
Smoking Cessation
Smoking cessation is the single most important modifiable risk factor in ILD and should be strongly encouraged in all current smokers at every clinical encounter. Smoking is the primary etiologic factor in respiratory bronchiolitis–associated ILD, desquamative interstitial pneumonia, and pulmonary Langerhans cell histiocytosis, all of which may stabilize or improve with cessation alone. In IPF and other fibrotic ILDs, smoking accelerates disease progression. Evidence-based cessation strategies, including pharmacotherapy (varenicline, bupropion, nicotine replacement therapy) and behavioral counseling, should be offered.
Antigen Avoidance in Hypersensitivity Pneumonitis
In hypersensitivity pneumonitis, identifying and avoiding the causative antigen is the cornerstone of management. Patients should be educated on the following points:
- Acute and subacute hypersensitivity pneumonitis: Complete and permanent antigen avoidance can lead to near-complete clinical resolution. Partial avoidance is insufficient; even occasional antigen exposure can perpetuate inflammation.
- Environmental modification: Modification may involve changes to the home environment (mold remediation, removal of avian pets, high-efficiency particulate air filtration), occupational adjustments (respiratory protection, workplace environmental assessment), or, in some cases, a change of occupation.
- Bird fancier lung: Patients must be counseled that rehoming their birds is often necessary because continued contact with avian antigens, despite medical treatment, leads to irreversible fibrosis in chronic hypersensitivity pneumonitis.
Occupational Exposure Avoidance
Patients with occupational ILD (silicosis, asbestosis, coal workers' pneumoconiosis) should be educated about the permanent nature of fibrotic lung disease and the importance of preventing further progression through complete cessation of occupational exposure. Appropriate respiratory protective equipment should be used for any residual workplace exposure. Occupational health referral and workers' compensation assessment may be appropriate.
Medication Awareness and Drug-ILD
Patients receiving medications known to cause ILD (amiodarone, methotrexate, nitrofurantoin, and immune checkpoint inhibitors) should be educated to report new or worsening dyspnea promptly. Patients should be advised not to discontinue medications without medical guidance, as in some cases the drug may be essential (eg, amiodarone for life-threatening arrhythmia) and the risk-benefit balance must be weighed by the prescribing clinician. Patients receiving immune checkpoint inhibitor therapy for cancer should be specifically counseled about the signs and symptoms of pneumonitis and instructed to present urgently if new dyspnea develops.
Vaccination
All patients with ILD should receive annual influenza and pneumococcal vaccinations (23-valent pneumococcal polysaccharide vaccine [PPSV23] and 20-valent pneumococcal conjugate vaccine [PCV20], per current Advisory Committee on Immunization Practices recommendations). Patients receiving immunosuppressive therapy should receive vaccinations before immunosuppression is initiated, where possible, as vaccine efficacy may be reduced during immunosuppression. Live vaccines are contraindicated in immunosuppressed patients
Exercise and Pulmonary Rehabilitation
Patients should be educated that regular physical activity and participation in pulmonary rehabilitation improve exercise tolerance, dyspnea, and quality of life, even when these interventions cannot reverse the underlying lung disease. A common misconception is that exercise is harmful in ILD; however, patients should be reassured that supervised exercise is safe and beneficial. Supplemental oxygen should be used during exercise if SpO2 falls below 88%.
Oxygen Therapy Education
Patients requiring supplemental oxygen should be educated on correct flow rates (rest versus activity versus sleep may require different settings), care of oxygen equipment, oxygen safety (no smoking near oxygen equipment; hazard of oxygen enrichment in enclosed spaces), and travel with oxygen (airline prenotification requirements; portable oxygen concentrator use)
Advance Care Planning
Given the progressive and life-limiting nature of ILD, particularly IPF, advance care planning should be introduced early, ideally at the time of diagnosis, and revisited at each clinical deterioration. Patients should be educated about disease trajectory, the limited reversibility of fibrosis, potential need for lung transplant evaluation, and end-of-life care preferences, including intubation and mechanical ventilation (generally not recommended in advanced IPF) and hospice care. Written advance directives should be completed and accessible to all treating clinicians.
Pearls and Other Issues
The following are clinical pearls about ILD:
- UIP pattern on HRCT means IPF until proven otherwise; always exclude CTD first. The hallmarks of UIP are subpleural, basal-predominant honeycombing ± traction bronchiectasis, temporal and spatial heterogeneity, and minimal ground glass opacities. When a confident UIP pattern is present on HRCT in the right clinical context, surgical biopsy is not required to diagnose IPF. However, the most important step before diagnosing a patient with IPF is to exclude CTD, particularly SSc, RA, and antisynthetase syndrome, as CTD-ILD with a UIP pattern is managed very differently (immunosuppression versus antifibrotic therapy).
- Velcro crackles and clubbing in a patient older than 50 years with a smoking history are consistent with IPF until proven otherwise. Fine, end-inspiratory, bibasilar crackles that sound like Velcro tearing, combined with digital clubbing (present in approximately 50% of patients with IPF), are the classic physical examination findings of IPF. In a patient older than 50 years who smokes or has smoked, this combination should trigger urgent HRCT and ILD specialist referral. Digital clubbing is uncommon in sarcoidosis and hypersensitivity pneumonitis; its presence supports a fibrotic pattern such as UIP.
- Anti–MDA5 antibody positivity indicates a rapidly progressive ILD emergency. Dermatomyositis-associated ILD with anti–MDA5 antibody is one of the few ILD emergencies. Unlike most CTD-ILD, which progresses slowly, anti–MDA5-positive dermatomyositis-ILD can progress to respiratory failure within weeks to months. Clinical recognition of clinically amyopathic dermatomyositis (skin but no muscle disease), mechanic hands, palmar papules, and bilateral ground-glass opacity on HRCT should prompt immediate anti–MDA5 testing and aggressive immunosuppression (combined corticosteroids, calcineurin inhibitor, and rituximab or cyclophosphamide). Missing this diagnosis can be fatal.
- Fibrotic hypersensitivity pneumonitis is the most common ILD to be misdiagnosed as IPF. Chronic fibrotic hypersensitivity pneumonitis can have a UIP-like pattern on HRCT and histology, leading to misdiagnosis as IPF. Key distinguishing features of fibrotic hypersensitivity pneumonitis include upper- or middle-lobe predominance (versus lower lobe predominance in IPF); lobular air trapping and mosaic attenuation on expiratory CT; poorly formed granulomas or giant cells on biopsy; antigen history (birds, mold, occupational); and BAL lymphocytosis greater than 20% to 30%. This distinction matters: fibrotic hypersensitivity pneumonitis may respond to antigen avoidance and immunosuppression, whereas IPF is treated with antifibrotic therapy.
- Tocilizumab is not a standard treatment for SSc-ILD. Despite being listed in some secondary sources, tocilizumab did not meet the primary endpoint (skin score) in the focuSSced phase 3 trial, and the FVC attenuation data were a secondary endpoint that did not reach statistical significance. Tocilizumab is not United States Food and Drug Administration (FDA)-approved for SSc-ILD and is not a guideline-recommended treatment. The correct first-line therapy for SSc-ILD is mycophenolate mofetil (stabilizes lung function over 52 weeks) or nintedanib (SENSCIS trial; FDA-approved 2019 for SSc-ILD
- Refer patients for lung transplant at diagnosis of IPF, not at end stage. The median waitlist time for a lung transplant is 2 to 4 years. A patient who deteriorates to end-stage IPF before referral may not survive long enough to receive a transplant. ATS and the International Society for Heart and Lung Transplantation guidelines recommend referral for transplant evaluation at the time of IPF diagnosis or at the first sign of physiological progression, so that workup can proceed in parallel with medical treatment. A bilateral lung transplant is preferred over a single-lung transplant in IPF.
- INBUILD trial: nintedanib works across all progressive fibrotic ILDs, not just IPF. The INBUILD trial established that nintedanib slows FVC decline in progressive pulmonary fibrosis of diverse etiology (fibrotic HP, RA-ILD, unclassifiable ILD, SSc-ILD with PPF features), not just IPF. Progressive pulmonary fibrosis is defined by meeting at least 2 of 3 criteria within 24 months: symptomatic worsening, radiological progression, or physiological decline (FVC decline of 5% or greater or DLCO decline of 10% or greater). When a patient with non-IPF ILD meets PPF criteria, nintedanib should be considered regardless of the underlying etiology.
Enhancing Healthcare Team Outcomes
The diagnosis and treatment of interstitial lung disease are complex and require an interdisciplinary team that includes the primary clinician, nurse practitioner, pulmonologist, thoracic surgeon, pathologist, and radiologist. Once the diagnosis is made, asymptomatic patients may be monitored, but all symptomatic patients need treatment. The condition is known to progress to fibrosis and end-stage lung disease; therefore, long-term monitoring is essential. Educating patients about the importance of smoking cessation is critical. The quality of life of most patients is poor, marked by significant respiratory distress with minimal physical activity.[61][89] Interstitial lung disease is best addressed by an interdisciplinary team that includes the patient's primary care clinician, a pulmonologist, nursing staff, and pharmacists, as well as pulmonary function technicians and respiratory therapists, to provide optimal patient diagnosis and care.
Media
(Click Image to Enlarge)
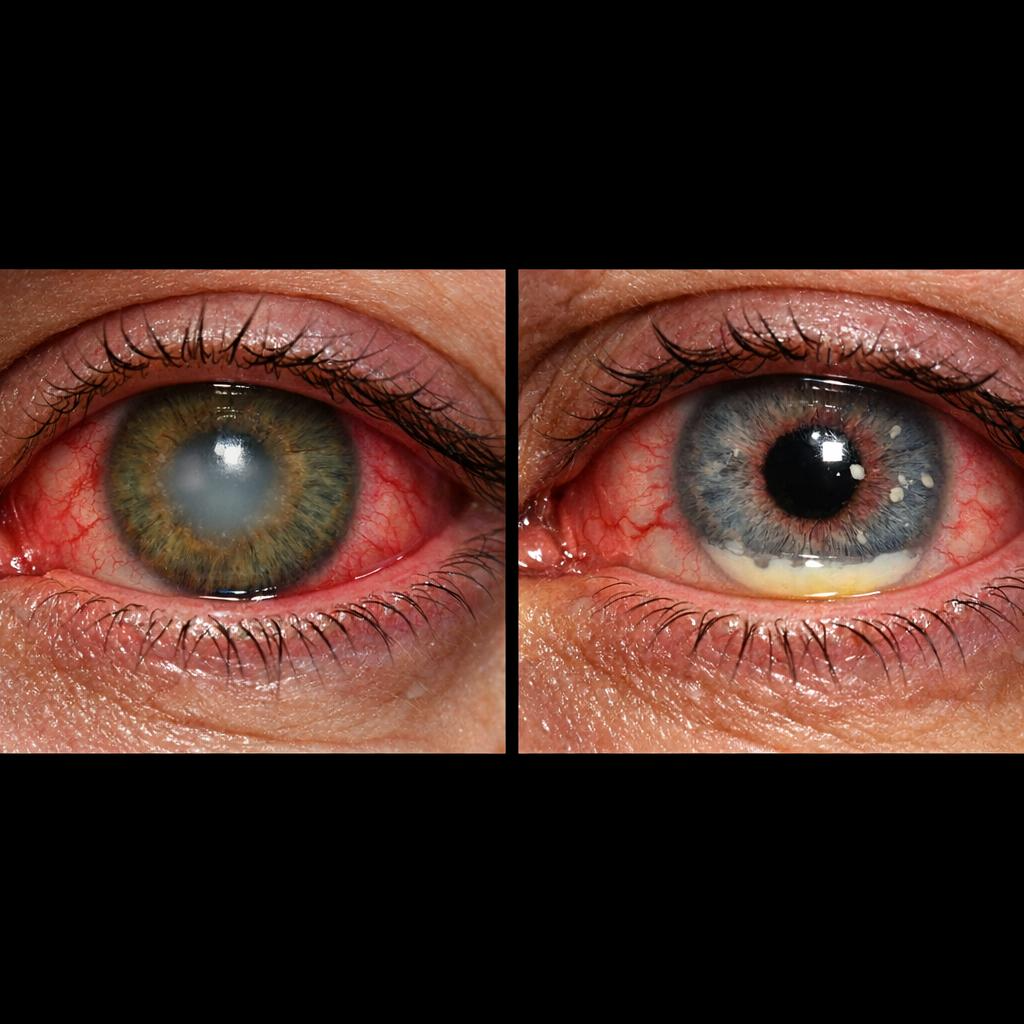
Uveitis Associated With Interstitial Lung Disease. Erythema surrounding the iris (left) and a collection of purulent material known as a hypopyon (right) can be signs of uveitis.
Contributed by StatPearls
(Click Image to Enlarge)
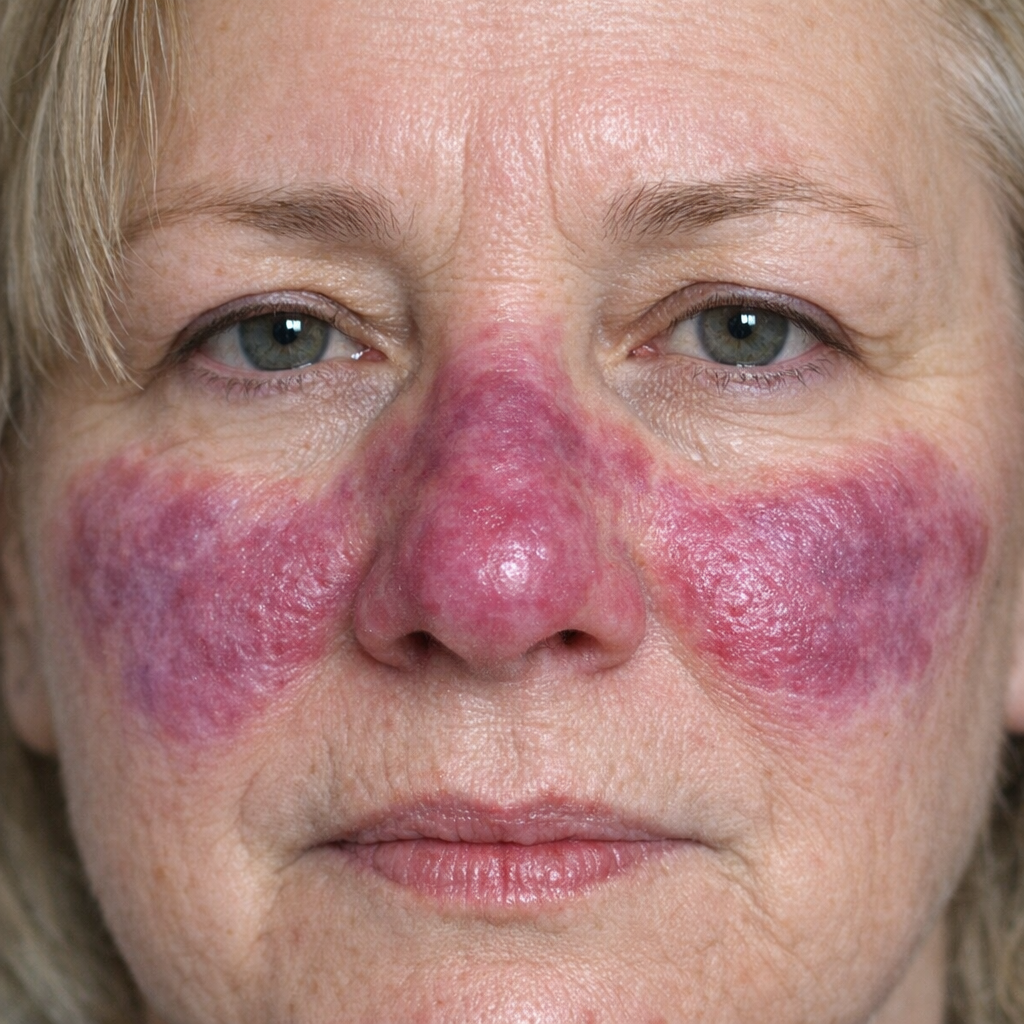
Lupus Pérnio. Violaceous plaques on the nose and cheeks can be a manifestation of sarcoidosis.
Contributed by StatPearls
(Click Image to Enlarge)
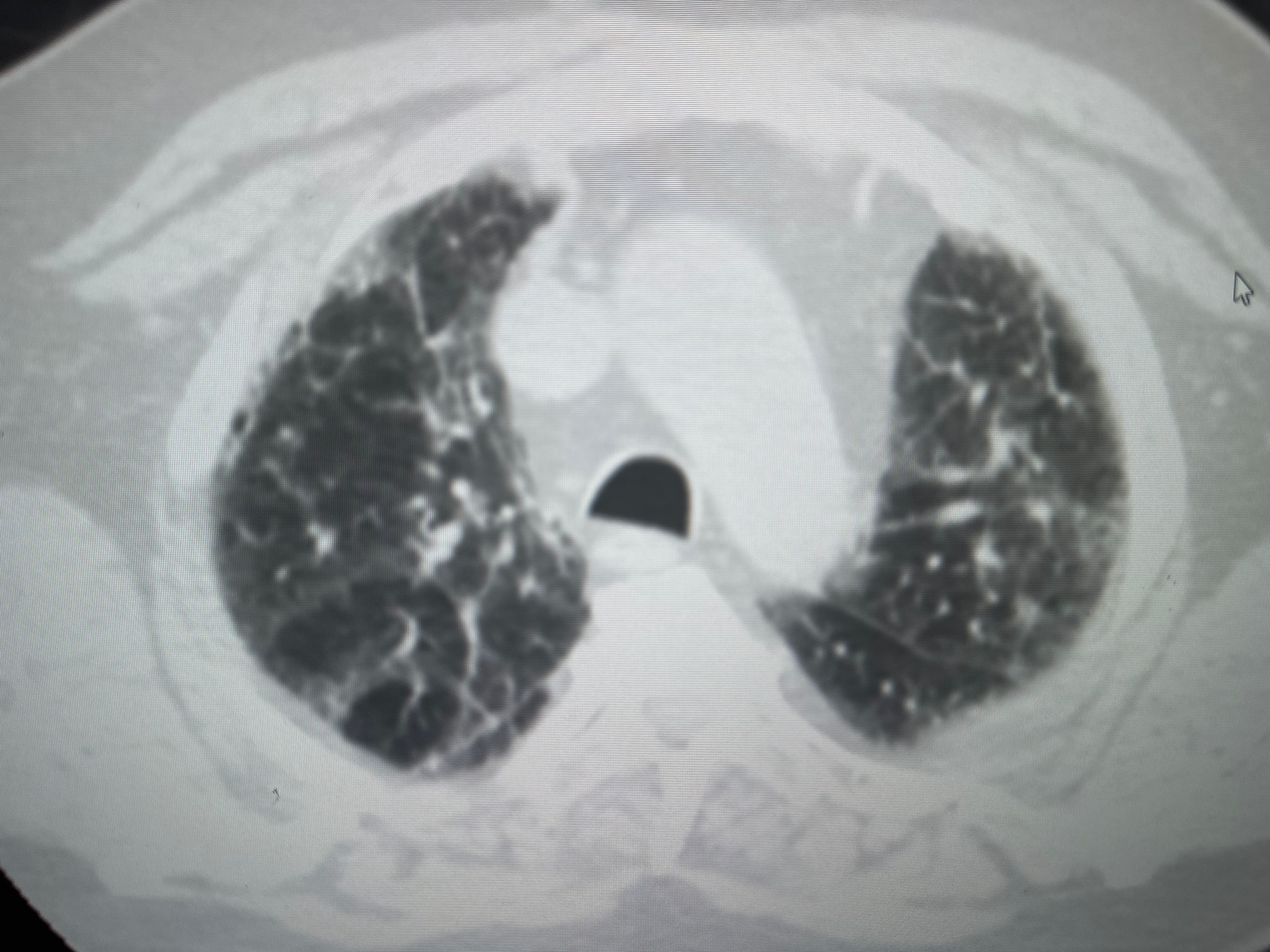
Usual Interstitial Pneumonia. This computed tomography scan demonstrates mid- and lower-lung predominant subpleural reticulation, atelectasis, and associated ground-glass opacities.
Contributed by A Sankari, MD, PhD
(Click Image to Enlarge)
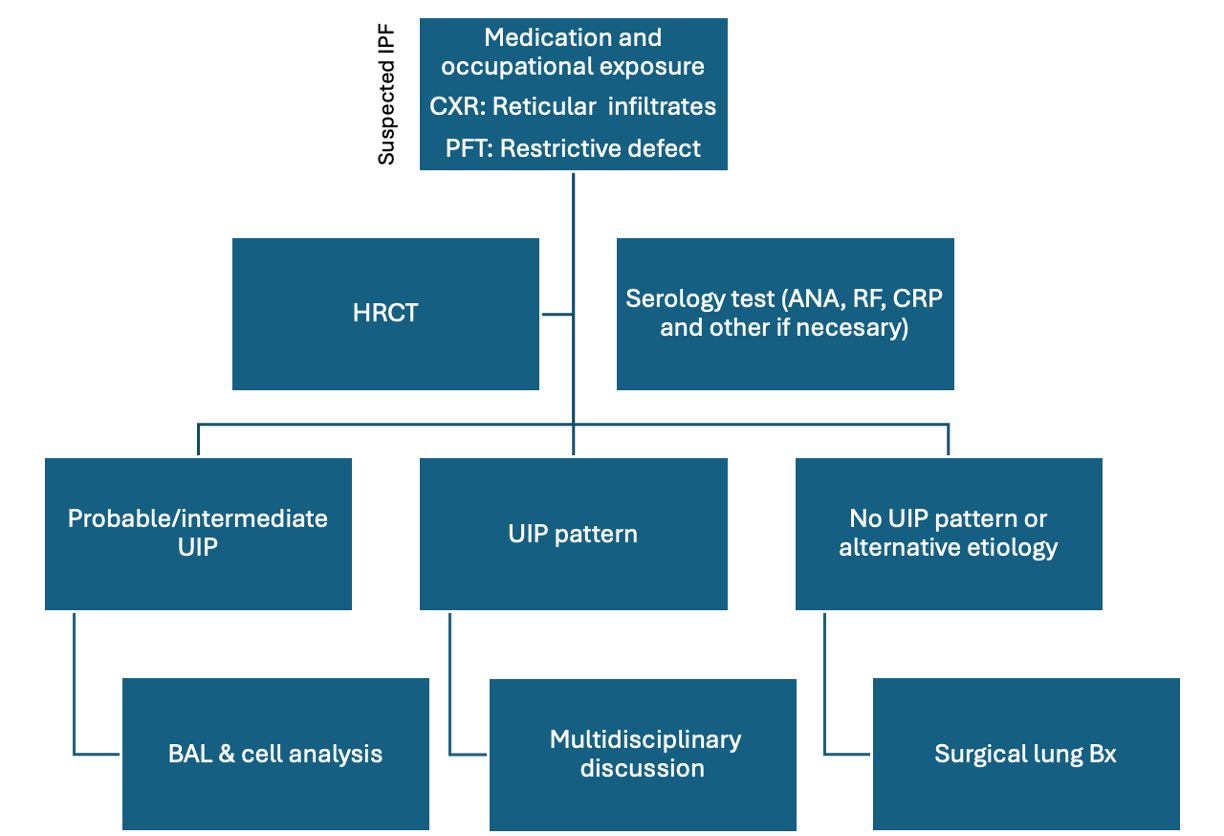
Idiopathic Pulmonary Fibrosis. The diagnostic algorithm for suspected idiopathic pulmonary fibrosis includes antibody titers, imaging, and tissue sampling.
Abbreviations: ANA, antinuclear antibodies; BAL, bronchoalveolar lavage; Bx, biopsy; CRP, C-reactive protein; CXR, chest x-ray; HRCT, high-resolution computed tomography scan; PFT, pulmonary function tests; RF, rheumatoid factor; UIP, usual interstitial pneumonia.
Contributed by A Sankari, MD, PhD
(Click Image to Enlarge)
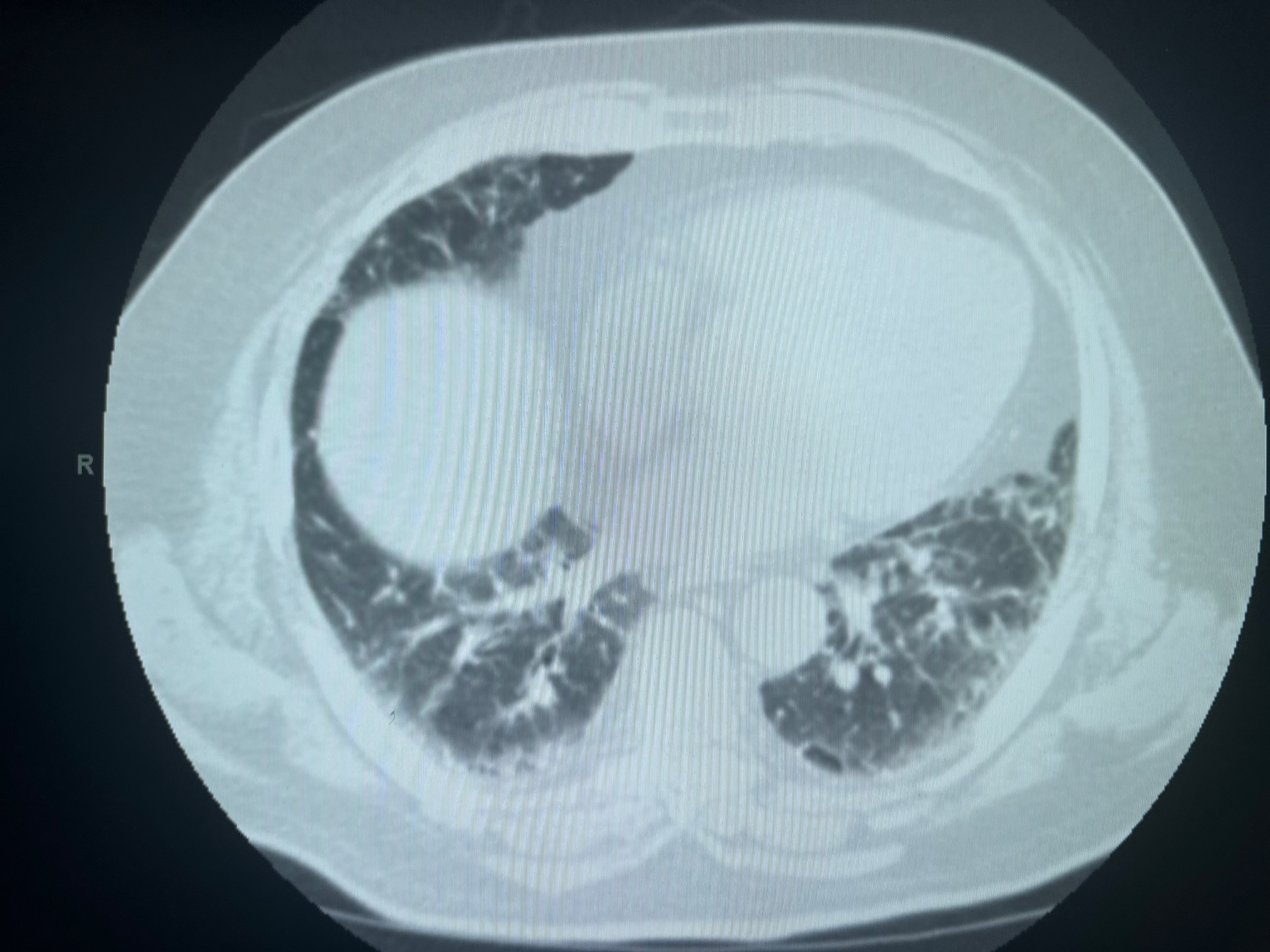
Computed Tomography of the Lungs in Usual Interstitial Pneumonia. Lower-lung predominant subpleural reticulation accompanied by ground-glass opacities or atelectasis is characteristic of usual interstitial pneumonia.
Contributed by A Sankari, MD, PhD
(Click Image to Enlarge)
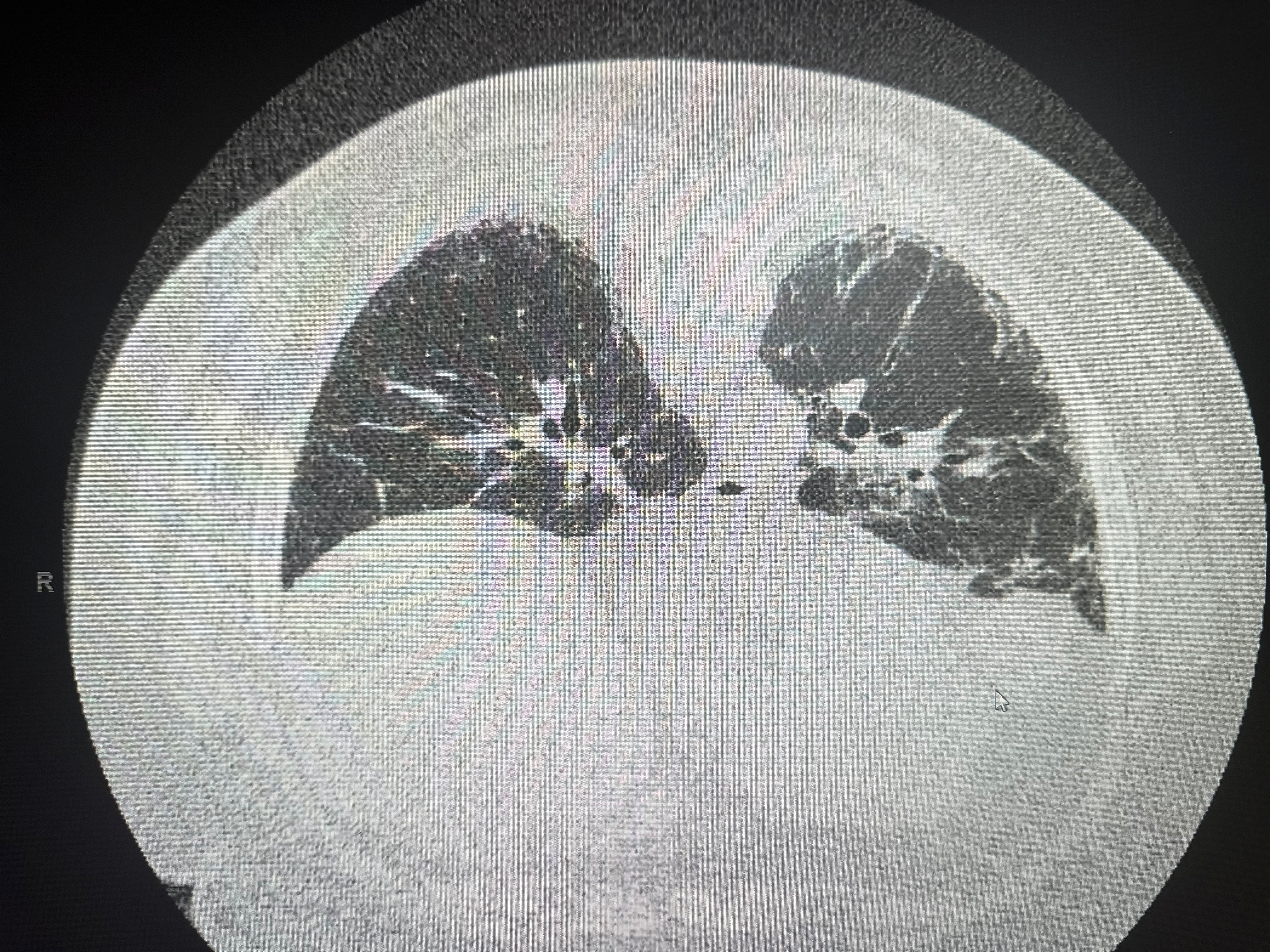
Usual Interstitial Pneumonia on Axial High-Resolution Computed Tomography. Subpleural, basal-predominant reticular opacities, traction bronchiectasis, and honeycombing are characteristic of usual interstitial pneumonia on high-resolution CT scan.
Contributed by A Sankari, MD, PhD
(Click Image to Enlarge)
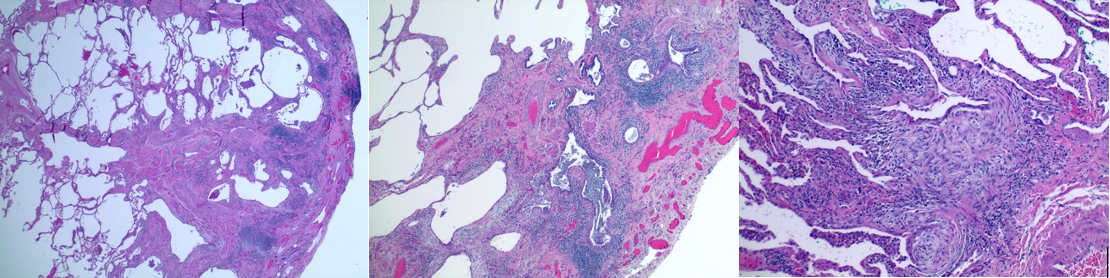
Histopathology of Usual Interstitial Pneumonia in Idiopathic Pulmonary Fibrosis. This image shows hematoxylin and eosin staining at magnifications of ×2, ×4, and ×10. The classic honeycombing changes in lung architecture, accompanied by an inflammatory cell infiltrate and a predominance of fibroblasts (blue), are consistent with usual interstitial pneumonia.
Contributed by A Sankari, MD, PhD
(Click Image to Enlarge)
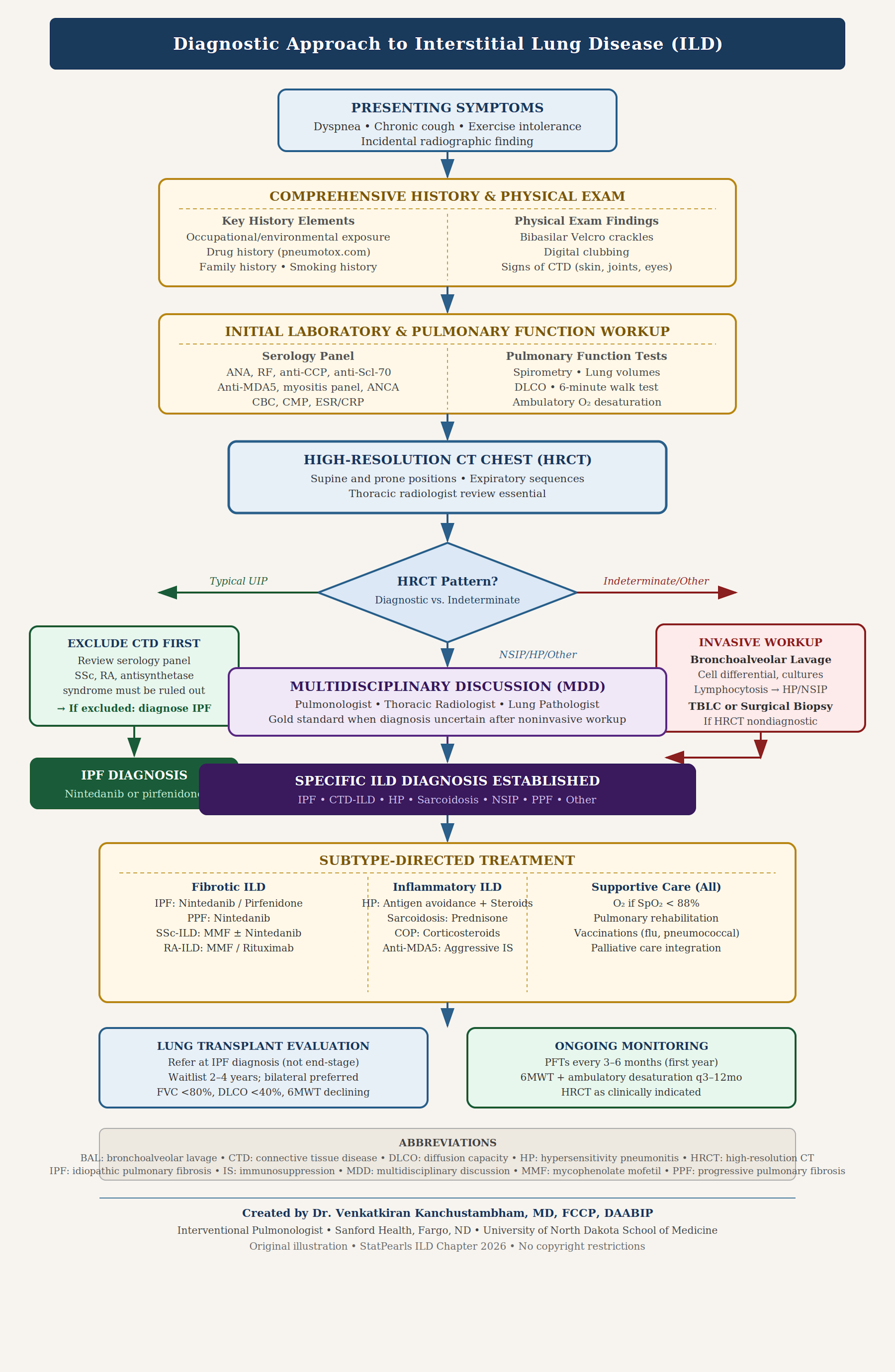
Diagnosis of Interstitial Lung Disease. An algorithm for diagnosis proceeds stepwise from initial presentation to history, pulmonary function testing, high-resolution computed tomography, interdisciplinary discussion, and subtype-directed treatment.
Created and contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)

High-Resolution Computed Tomography Patterns of Interstitial Lung Disease. 1. Subpleural basal honeycombing and traction bronchiectasis are characteristic of usual interstitial pneumonia. 2. Bilateral ground-glass opacity and relative subpleural sparing are common in nonspecific interstitial pneumonia. 3. Mosaic attenuation in the upper and middle lobes is associated with chronic fibrotic hypersensitivity pneumonitis. 4. Sarcoidosis often demonstrates bilateral hilar adenopathy and perilymphatic nodules.
Created and contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)

Clinical Examination Findings of Interstitial Lung Disease. This image shows the following: 1. Fine-end-inspiratory bibasilar crackles are present in 80% of patients with ILD; 2. Digital clubbing may be apparent with the Schamroth window test; 3. Mechanic hands and Gottron papules may indicate dermatomyositis. Lupus pernio is associated with sarcoidosis; and 4. Signs of advanced disease include pulmonary hypertension, hypoxemia, cachexia, and cor pulmonale.
Created and contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)

Etiologies of Interstitial Lung Disease. The etiologies of interstitial lung disease can be categorized into 5 groups: idiopathic, connective tissue disease-associated, granulomatous, environmental/occupational, and drug-induced.
Created and contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)

Drug-Induced Interstitial Lung Disease. Direct cytotoxicity, immune-mediated hypersensitivity, phospholipidosis, and vascular toxicity are mechanisms associated with drug-induced interstitial lung disease. Clinical pearls in diagnosis and treatment are also presented.
Created and contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)
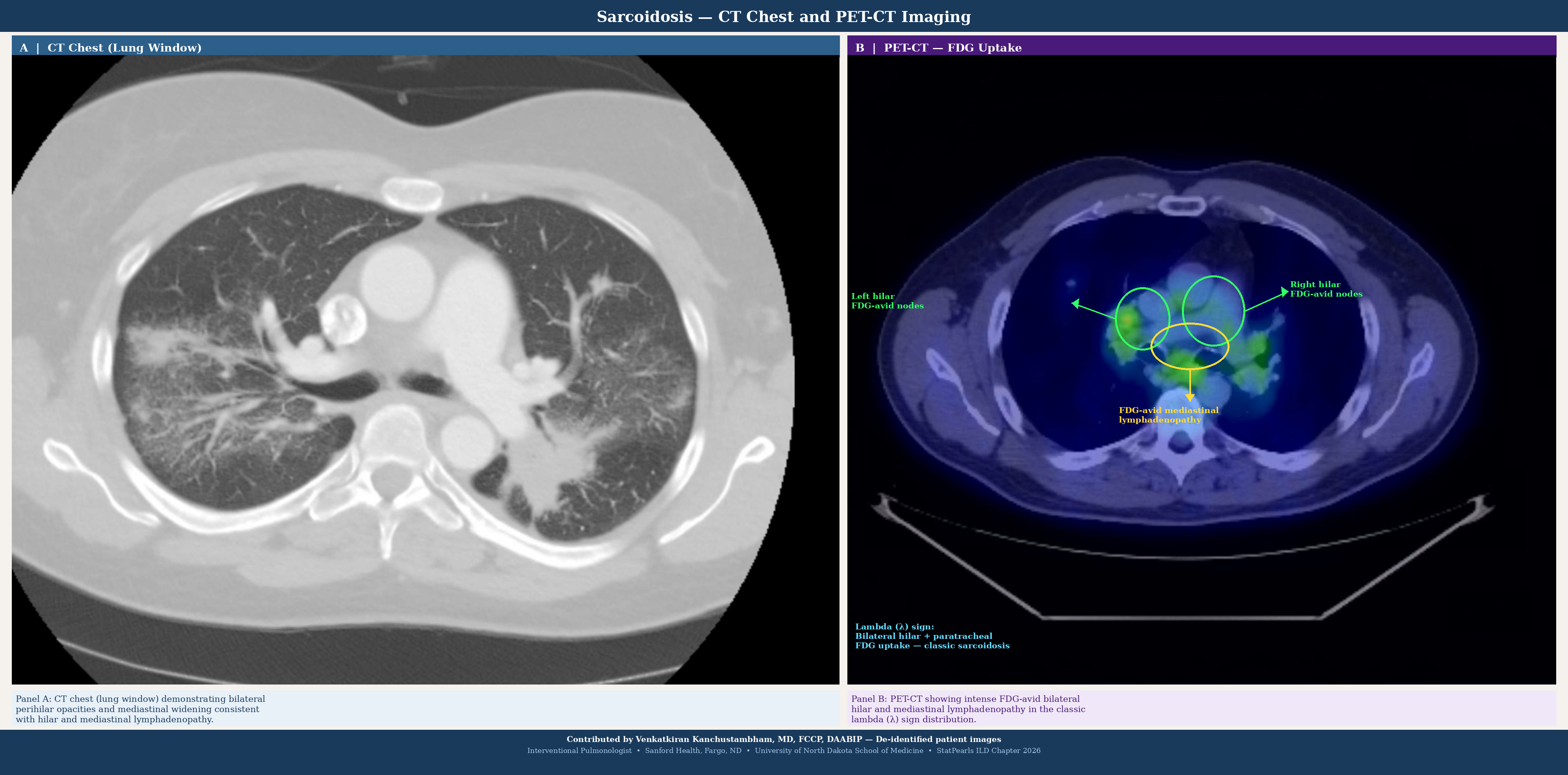
Sarcoidosis Imaging. Panel A shows computed tomography of the chest with bilateral perilymphatic nodules along bronchovascular bundles and bilateral hilar and mediastinal adenopathy. Panel B shows positron-emission tomography with hyperintensity around bilateral hilar and mediastinal lymph nodes with a classic ƛ sign distribution.
Contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)
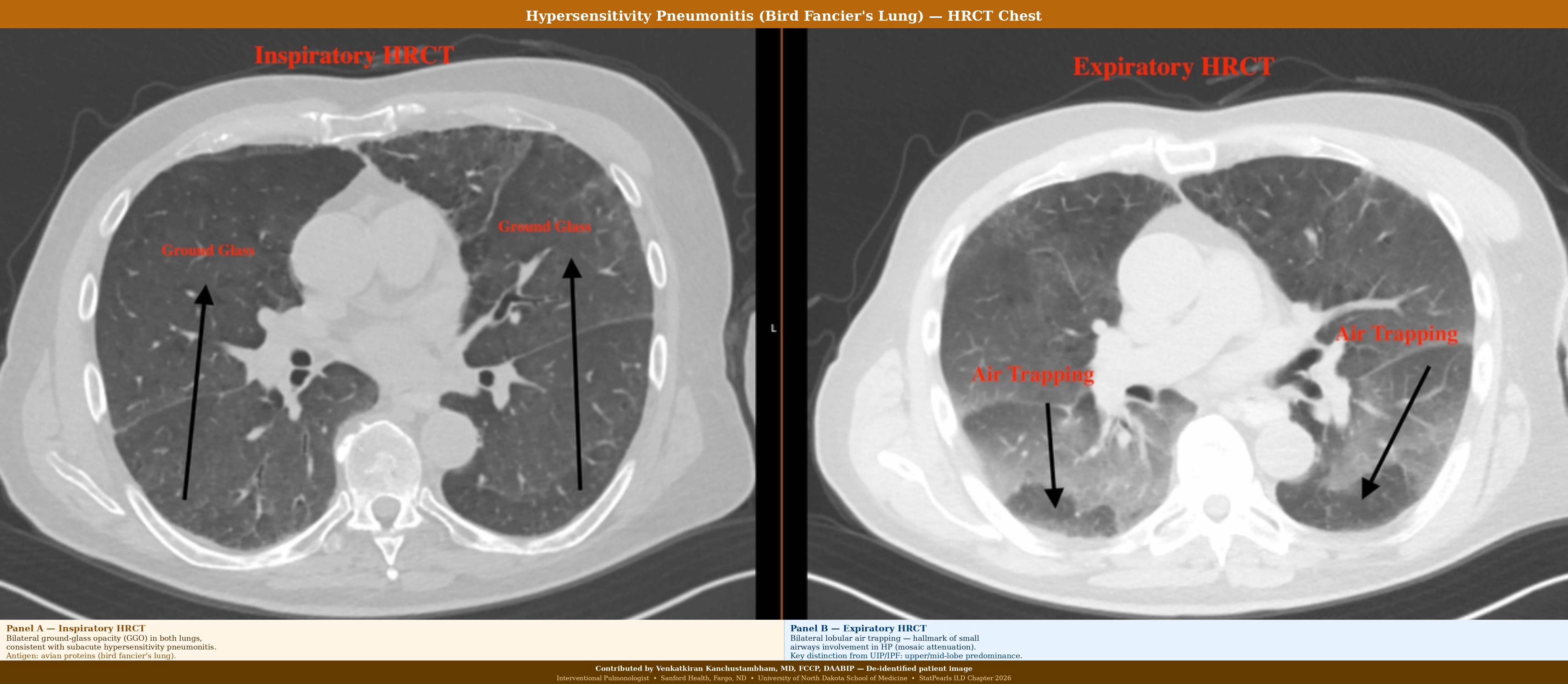
Hypersensitivity Pneumonitis. Panel A shows bilateral ground-glass opacities on high-resolution computed tomography. Panel B shows a high-resolution computed tomography with bilateral lobular air trapping in the upper and middle lobes.
Contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)
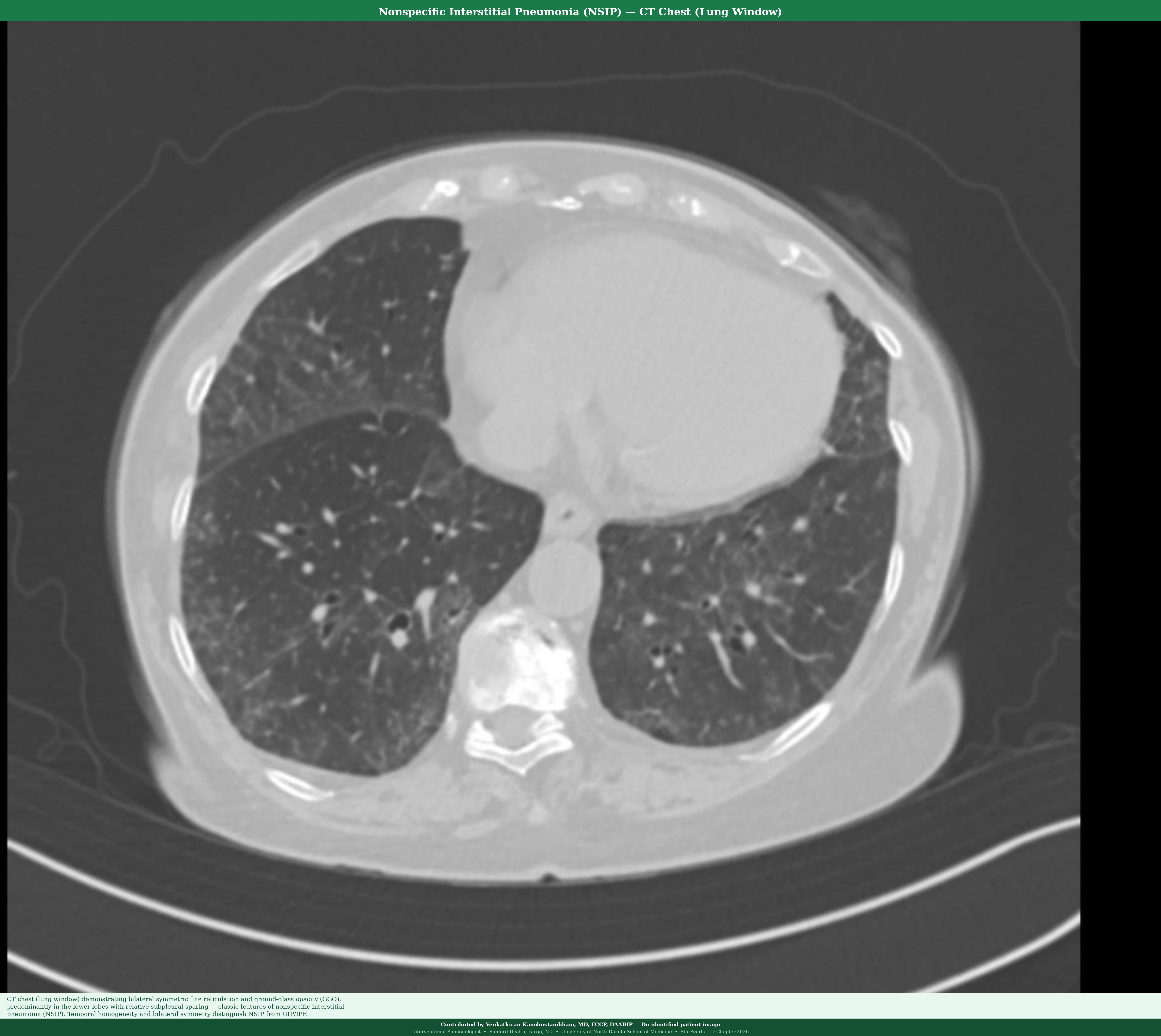
Nonspecific Interstitial Pneumonia. Bilateral, symmetric, fine reticulation and ground-glass opacities predominate in the lower lobes with subpleural sparing on the computed tomography scan. The temporal homogeneity and bilateral symmetry distinguish nonspecific interstitial pneumonia from usual interstitial pneumonia.
Contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)
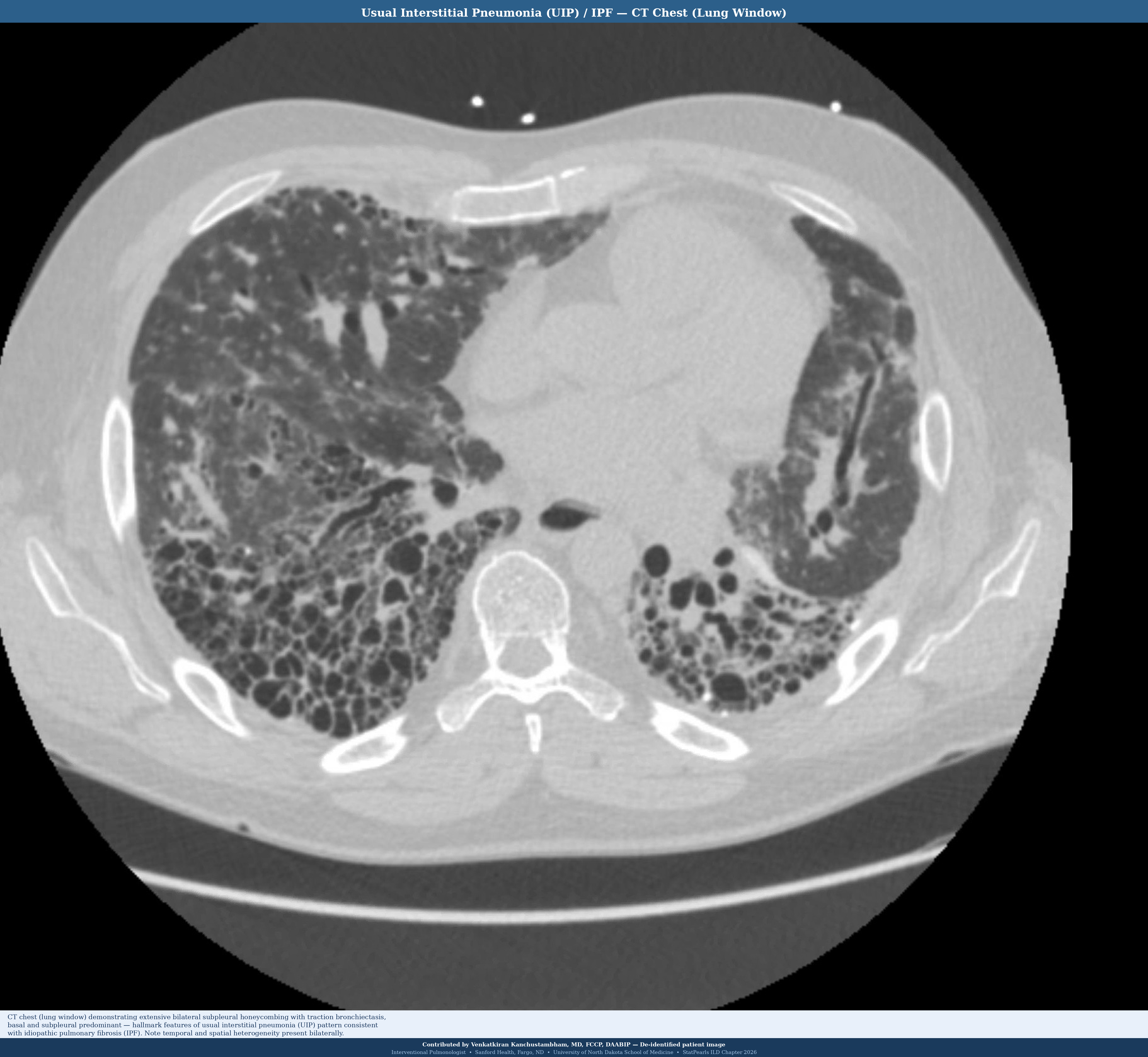
Pattern of Usual Interstitial Pneumonia/ Idiopathic Pulmonary Fibrosis on Computed Tomography Scan. Extensive bilateral subpleural honeycombing with traction bronchiectasis, predominant in the basal and subpleural areas. Bilateral, temporal, and spatial heterogeneity are features consistent with usual interstitial pneumonia.
Contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)
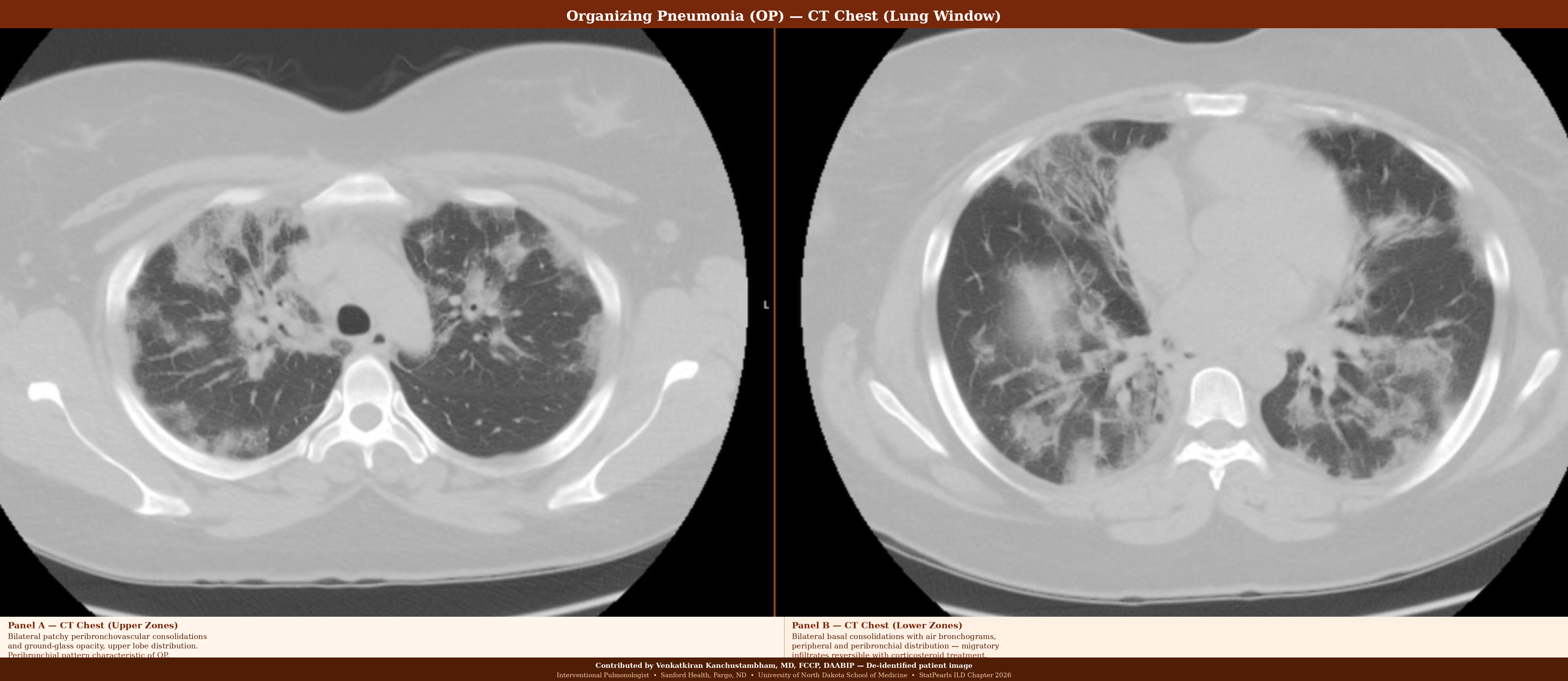
Organizing Pneumonia. Panel A shows bilateral patchy peribronchovascular consolidation and ground-glass opacity in the upper lobes on chest computed tomography. Panel B shows bilateral basal consolidation with air bronchograms in peripheral and peribronchial areas, along with migratory infiltrates characteristic of organizing pneumonia.
Contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)
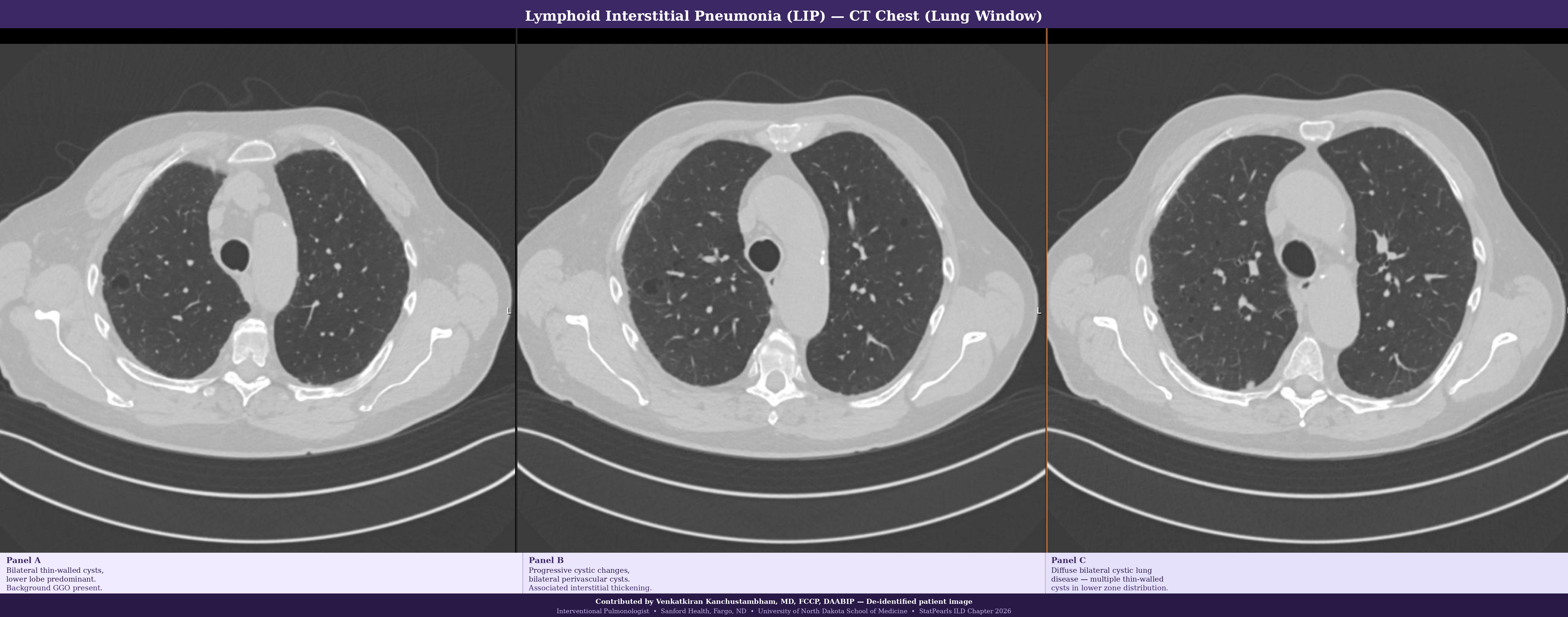
Lymphoid Interstitial Pneumonia. Bilateral thin-walled cysts and ground-glass opacities are seen in the lower lobes on this chest computed tomography, characteristic of lymphoid interstitial pneumonia.
Contributed by V Kanchustambham, MD, FCCP, CAQ
(Click Image to Enlarge)
Diffuse Interstitial Lung Disease. Two axial computed tomography images at the level of the lung base demonstrate diffuse bilateral reticulonodular opacities and honeycombing characteristic of interstitial lung disease.
Contributed by D Tafti, MD
References
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ, ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. American journal of respiratory and critical care medicine. 2011 Mar 15:183(6):788-824. doi: 10.1164/rccm.2009-040GL. Epub [PubMed PMID: 21471066]
Level 1 (high-level) evidenceRivera-Ortega P, Molina-Molina M. Interstitial Lung Diseases in Developing Countries. Annals of global health. 2019 Jan 22:85(1):. pii: 4. doi: 10.5334/aogh.2414. Epub 2019 Jan 22 [PubMed PMID: 30741505]
Mikolasch TA, Garthwaite HS, Porter JC. Update in diagnosis and management of interstitial lung disease . Clinical medicine (London, England). 2017 Apr:17(2):146-153. doi: 10.7861/clinmedicine.17-2-146. Epub [PubMed PMID: 28365626]
Ryerson CJ, Adegunsoye A, Piciucchi S, Hariri LP, Khor YH, Wijsenbeek MS, Wells AU, Sharma A, Cooper WA, Antoniou K, Borie R, Fabre A, Inoue Y, Johannson K, Johkoh T, Kawano-Dourado L, Kazerooni E, Maher TM, Molyneaux PL, Protti R, Ravaglia C, Renzoni EA, Saito-Koyama R, Sverzellati N, Walsh SLF, Wolters P, Yang SR, Travis W, Nicholson AG. Update of the international multidisciplinary classification of the interstitial pneumonias: an ERS/ATS statement. The European respiratory journal. 2025 Dec:66(6):. pii: 2500158. doi: 10.1183/13993003.00158-2025. Epub 2025 Dec 4 [PubMed PMID: 40774805]
Capron F. [New classification of interstitial lung disease]. Revue de pneumologie clinique. 2005 Jun:61(3):133-40 [PubMed PMID: 16142185]
Luckhardt TR, Müller-Quernheim J, Thannickal VJ. Update in diffuse parenchymal lung disease 2011. American journal of respiratory and critical care medicine. 2012 Jul 1:186(1):24-9. doi: 10.1164/rccm.201203-0509UP. Epub [PubMed PMID: 22753686]
Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. American journal of respiratory and critical care medicine. 2017 Sep 15:196(6):680-689. doi: 10.1164/rccm.201611-2201PP. Epub [PubMed PMID: 28598197]
Level 3 (low-level) evidenceMira-Avendano I, Abril A, Burger CD, Dellaripa PF, Fischer A, Gotway MB, Lee AS, Lee JS, Matteson EL, Yi ES, Ryu JH. Interstitial Lung Disease and Other Pulmonary Manifestations in Connective Tissue Diseases. Mayo Clinic proceedings. 2019 Feb:94(2):309-325. doi: 10.1016/j.mayocp.2018.09.002. Epub 2018 Dec 14 [PubMed PMID: 30558827]
Kalchiem-Dekel O, Galvin JR, Burke AP, Atamas SP, Todd NW. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. Journal of clinical medicine. 2018 Nov 24:7(12):. doi: 10.3390/jcm7120476. Epub 2018 Nov 24 [PubMed PMID: 30477216]
Schwaiblmair M, Behr W, Haeckel T, Märkl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. The open respiratory medicine journal. 2012:6():63-74. doi: 10.2174/1874306401206010063. Epub 2012 Jul 27 [PubMed PMID: 22896776]
Skeoch S, Weatherley N, Swift AJ, Oldroyd A, Johns C, Hayton C, Giollo A, Wild JM, Waterton JC, Buch M, Linton K, Bruce IN, Leonard C, Bianchi S, Chaudhuri N. Drug-Induced Interstitial Lung Disease: A Systematic Review. Journal of clinical medicine. 2018 Oct 15:7(10):. doi: 10.3390/jcm7100356. Epub 2018 Oct 15 [PubMed PMID: 30326612]
Level 1 (high-level) evidenceHortense AB, Santos MKD, Wada D, Fabro AT, Lima M, Rodrigues S, Calado RT, Baddini-Martinez J. Familial pulmonary fibrosis: a heterogeneous spectrum of presentations. Jornal brasileiro de pneumologia : publicacao oficial da Sociedade Brasileira de Pneumologia e Tisilogia. 2019 Jun 10:45(5):e20180079. doi: 10.1590/1806-3713/e20180079. Epub 2019 Jun 10 [PubMed PMID: 31188976]
Raghu G, Nyberg F, Morgan G. The epidemiology of interstitial lung disease and its association with lung cancer. British journal of cancer. 2004 Aug:91 Suppl 2(Suppl 2):S3-10 [PubMed PMID: 15340372]
Maher TM. Interstitial Lung Disease: A Review. JAMA. 2024 May 21:331(19):1655-1665. doi: 10.1001/jama.2024.3669. Epub [PubMed PMID: 38648021]
Spagnolo P, Guler SA, Chaudhuri N, Udwadia Z, Sesé L, Kaul B, Enghelmayer JI, Valenzuela C, Malhotra A, Ryerson CJ, Khor YH, Corte TJ, Cottin V. Global epidemiology and burden of interstitial lung disease. The Lancet. Respiratory medicine. 2025 Aug:13(8):739-755. doi: 10.1016/S2213-2600(25)00129-8. Epub 2025 Jul 17 [PubMed PMID: 40684782]
Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. European respiratory review : an official journal of the European Respiratory Society. 2012 Dec 1:21(126):355-61. doi: 10.1183/09059180.00002512. Epub [PubMed PMID: 23204124]
Kaul B, Cottin V, Collard HR, Valenzuela C. Variability in Global Prevalence of Interstitial Lung Disease. Frontiers in medicine. 2021:8():751181. doi: 10.3389/fmed.2021.751181. Epub 2021 Nov 4 [PubMed PMID: 34805219]
Joy GM, Arbiv OA, Wong CK, Lok SD, Adderley NA, Dobosz KM, Johannson KA, Ryerson CJ. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. European respiratory review : an official journal of the European Respiratory Society. 2023 Mar 31:32(167):. doi: 10.1183/16000617.0210-2022. Epub 2023 Mar 8 [PubMed PMID: 36889782]
Level 1 (high-level) evidenceOlson A, Hartmann N, Patnaik P, Wallace L, Schlenker-Herceg R, Nasser M, Richeldi L, Hoffmann-Vold AM, Cottin V. Estimation of the Prevalence of Progressive Fibrosing Interstitial Lung Diseases: Systematic Literature Review and Data from a Physician Survey. Advances in therapy. 2021 Feb:38(2):854-867. doi: 10.1007/s12325-020-01578-6. Epub 2020 Dec 14 [PubMed PMID: 33315170]
Level 1 (high-level) evidenceHoffmann-Vold AM, Maher TM, Philpot EE, Ashrafzadeh A, Barake R, Barsotti S, Bruni C, Carducci P, Carreira PE, Castellví I, Del Galdo F, Distler JHW, Foeldvari I, Fraticelli P, George PM, Griffiths B, Guillén-Del-Castillo A, Hamid AM, Horváth R, Hughes M, Kreuter M, Moazedi-Fuerst F, Olas J, Paul S, Rotondo C, Rubio-Rivas M, Seferian A, Tomčík M, Uzunhan Y, Walker UA, Więsik-Szewczyk E, Distler O. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. The Lancet. Rheumatology. 2020 Feb:2(2):e71-e83. doi: 10.1016/S2665-9913(19)30144-4. Epub 2020 Jan 14 [PubMed PMID: 38263663]
Level 3 (low-level) evidenceSuki B, Stamenović D, Hubmayr R. Lung parenchymal mechanics. Comprehensive Physiology. 2011 Jul:1(3):1317-51. doi: 10.1002/cphy.c100033. Epub [PubMed PMID: 23733644]
Level 3 (low-level) evidenceSankari A, Chapman K, Ullah S. Idiopathic Pulmonary Fibrosis. StatPearls. 2026 Jan:(): [PubMed PMID: 28846333]
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina-Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia-Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022 May 1:205(9):e18-e47. doi: 10.1164/rccm.202202-0399ST. Epub [PubMed PMID: 35486072]
Level 1 (high-level) evidenceRaghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, Azuma A, Bice TJ, Bouros D, Brown KK, Collard HR, Duggal A, Galvin L, Inoue Y, Jenkins RG, Johkoh T, Kazerooni EA, Kitaichi M, Knight SL, Mansour G, Nicholson AG, Pipavath SNJ, Buendía-Roldán I, Selman M, Travis WD, Walsh S, Wilson KC, American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2018 Sep 1:198(5):e44-e68. doi: 10.1164/rccm.201807-1255ST. Epub [PubMed PMID: 30168753]
Level 1 (high-level) evidenceLange SM, Sankari A, Parekh M. Collagen Vascular Disease Associated With Interstitial Lung. StatPearls. 2026 Jan:(): [PubMed PMID: 32644520]
Antoniou KM, Distler O, Gheorghiu AM, Moor CC, Vikse J, Bizymi N, Galetti I, Brown G, Bargagli E, Allanore Y, Corte TJ, Dieudé P, Cottin V, Fisher BA, Fabre A, Giles JT, Kreuter M, Lundberg IE, Poletti V, Maurer B, Renzoni EA, Müller-Ladner U, Strek ME, Sverzellati N, Studenic P, Mohammed J, Nagavci B, Stamm T, Tonia T, Crestani B, Hoffmann-Vold AM. ERS/EULAR clinical practice guidelines for connective tissue disease-associated interstitial lung disease. The European respiratory journal. 2026 Jan:67(1):. doi: 10.1183/13993003.02533-2024. Epub 2026 Jan 22 [PubMed PMID: 40907995]
Level 1 (high-level) evidenceAntoniou KM, Distler O, Gheorghiu AM, Moor CC, Vikse J, Bizymi N, Galetti I, Brown G, Bargagli E, Allanore Y, Corte TJ, Dieudé P, Cottin V, Fisher BA, Fabre A, Giles JT, Kreuter M, Lundberg IE, Poletti V, Maurer B, Renzoni EA, Müller-Ladner U, Strek ME, Sverzellati N, Studenic P, Mohammed J, Nagavci B, Stamm T, Tonia T, Crestani B, Hoffmann-Vold AM. ERS/EULAR clinical practice guidelines for connective tissue disease-associated interstitial lung disease developed by the task force for connective tissue disease-associated interstitial lung disease of the European Respiratory Society (ERS) and the European Alliance of Associations for Rheumatology (EULAR) Endorsed by the European Reference Network on rare respiratory diseases (ERN-LUNG). Annals of the rheumatic diseases. 2026 Jan:85(1):22-60. doi: 10.1016/j.ard.2025.08.021. Epub 2025 Sep 4 [PubMed PMID: 40912974]
Level 1 (high-level) evidenceRaghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, Bargagli E, Chung JH, Collins BF, Bendstrup E, Chami HA, Chua AT, Corte TJ, Dalphin JC, Danoff SK, Diaz-Mendoza J, Duggal A, Egashira R, Ewing T, Gulati M, Inoue Y, Jenkins AR, Johannson KA, Johkoh T, Tamae-Kakazu M, Kitaichi M, Knight SL, Koschel D, Lederer DJ, Mageto Y, Maier LA, Matiz C, Morell F, Nicholson AG, Patolia S, Pereira CA, Renzoni EA, Salisbury ML, Selman M, Walsh SLF, Wuyts WA, Wilson KC. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2020 Aug 1:202(3):e36-e69. doi: 10.1164/rccm.202005-2032ST. Epub [PubMed PMID: 32706311]
Level 1 (high-level) evidenceChandra D, Cherian SV. Hypersensitivity Pneumonitis. StatPearls. 2026 Jan:(): [PubMed PMID: 29763093]
Blanc PD, Annesi-Maesano I, Balmes JR, Cummings KJ, Fishwick D, Miedinger D, Murgia N, Naidoo RN, Reynolds CJ, Sigsgaard T, Torén K, Vinnikov D, Redlich CA. The Occupational Burden of Nonmalignant Respiratory Diseases. An Official American Thoracic Society and European Respiratory Society Statement. American journal of respiratory and critical care medicine. 2019 Jun 1:199(11):1312-1334. doi: 10.1164/rccm.201904-0717ST. Epub [PubMed PMID: 31149852]
DeLight N, Sachs H. Pneumoconiosis. StatPearls. 2026 Jan:(): [PubMed PMID: 32310362]
Spagnolo P, Bonniaud P, Rossi G, Sverzellati N, Cottin V. Drug-induced interstitial lung disease. The European respiratory journal. 2022 Oct:60(4):. pii: 2102776. doi: 10.1183/13993003.02776-2021. Epub 2022 Oct 27 [PubMed PMID: 35332071]
Miedema JR, Bonella F, Buschulte K, Culver DA, Jeny F, Obi ON, Rivera NV, Spagnolo P, Veltkamp M, Wijsenbeek M. Sarcoidosis: a state-of-the-art review. The European respiratory journal. 2026 Feb:67(2):. pii: 2501324. doi: 10.1183/13993003.01324-2025. Epub 2026 Feb 19 [PubMed PMID: 41232941]
Sankari A, Basit H, Sharma S. Desquamative Interstitial Pneumonia. StatPearls. 2026 Jan:(): [PubMed PMID: 30252335]
Nayfeh AS, Chippa V, Moore DR. Nonspecific Interstitial Pneumonia. StatPearls. 2026 Jan:(): [PubMed PMID: 30085516]
Mrad A, Huda N. Acute Interstitial Pneumonia(Archived). StatPearls. 2026 Jan:(): [PubMed PMID: 32119316]
Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. American journal of respiratory and critical care medicine. 1998 Apr:157(4 Pt 1):1301-15 [PubMed PMID: 9563754]
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D, ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. American journal of respiratory and critical care medicine. 2013 Sep 15:188(6):733-48. doi: 10.1164/rccm.201308-1483ST. Epub [PubMed PMID: 24032382]
Cordier JF. Cryptogenic organising pneumonia. The European respiratory journal. 2006 Aug:28(2):422-46 [PubMed PMID: 16880372]
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, Lee JS, Maher TM, Wells AU, Antoniou KM, Behr J, Brown KK, Cottin V, Flaherty KR, Fukuoka J, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kolb M, Lynch DA, Myers JL, Raghu G, Richeldi L, Taniguchi H, Martinez FJ. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. American journal of respiratory and critical care medicine. 2016 Aug 1:194(3):265-75. doi: 10.1164/rccm.201604-0801CI. Epub [PubMed PMID: 27299520]
Margaritopoulos GA, Vasarmidi E, Jacob J, Wells AU, Antoniou KM. Smoking and interstitial lung diseases. European respiratory review : an official journal of the European Respiratory Society. 2015 Sep:24(137):428-35. doi: 10.1183/16000617.0050-2015. Epub [PubMed PMID: 26324804]
Lentz RJ, Argento AC, Colby TV, Rickman OB, Maldonado F. Transbronchial cryobiopsy for diffuse parenchymal lung disease: a state-of-the-art review of procedural techniques, current evidence, and future challenges. Journal of thoracic disease. 2017 Jul:9(7):2186-2203. doi: 10.21037/jtd.2017.06.96. Epub [PubMed PMID: 28840020]
Perrichon M, Coudert B, Foucher P, Bailly F, Solary E, Jeannin L, Camus P. [Diagnostic value of broncho-alveolar lavage in lung diseases caused by amiodarone]. Presse medicale (Paris, France : 1983). 1989 Mar 11:18(10):542 [PubMed PMID: 2523052]
Poveda JJR, Tamayo MH, Huizi JJZ, Sellares J, Hernández-González F. Drug-Induced Pulmonary Toxicity in the Era of Immunotherapy and Biologics: A Narrative Review of Mechanism, Diagnosis, and Management. Pulmonary therapy. 2026 Mar:12(1):99-116. doi: 10.1007/s41030-025-00338-7. Epub 2026 Jan 4 [PubMed PMID: 41484545]
Level 3 (low-level) evidenceCamus P, Bonniaud P, Fanton A, Camus C, Baudaun N, Foucher P. Drug-induced and iatrogenic infiltrative lung disease. Clinics in chest medicine. 2004 Sep:25(3):479-519, vi [PubMed PMID: 15331188]
Hnizdo E, Murray J. Risk of pulmonary tuberculosis relative to silicosis and exposure to silica dust in South African gold miners. Occupational and environmental medicine. 1998 Jul:55(7):496-502 [PubMed PMID: 9816385]
Nishino M, Giobbie-Hurder A, Hatabu H, Ramaiya NH, Hodi FS. Incidence of Programmed Cell Death 1 Inhibitor-Related Pneumonitis in Patients With Advanced Cancer: A Systematic Review and Meta-analysis. JAMA oncology. 2016 Dec 1:2(12):1607-1616. doi: 10.1001/jamaoncol.2016.2453. Epub [PubMed PMID: 27540850]
Level 1 (high-level) evidenceSève P, Pacheco Y, Durupt F, Jamilloux Y, Gerfaud-Valentin M, Isaac S, Boussel L, Calender A, Androdias G, Valeyre D, El Jammal T. Sarcoidosis: A Clinical Overview from Symptoms to Diagnosis. Cells. 2021 Mar 31:10(4):. doi: 10.3390/cells10040766. Epub 2021 Mar 31 [PubMed PMID: 33807303]
Level 3 (low-level) evidenceJakubczyc A, Neurohr C. [Diagnosis and Treatment of Interstitial Lung Diseases]. Deutsche medizinische Wochenschrift (1946). 2018 Dec:143(24):1774-1777. doi: 10.1055/a-0622-9299. Epub 2018 Dec 3 [PubMed PMID: 30508858]
Mehta AA, Paul T, Cb M, Haridas N. Anti-MDA5 antibody-positive dermatomyositis with rapidly progressive interstitial lung disease: report of two cases. BMJ case reports. 2021 Apr 28:14(4):. doi: 10.1136/bcr-2020-240046. Epub 2021 Apr 28 [PubMed PMID: 33910791]
Level 3 (low-level) evidenceRaghu G, Brown KK. Interstitial lung disease: clinical evaluation and keys to an accurate diagnosis. Clinics in chest medicine. 2004 Sep:25(3):409-19, v [PubMed PMID: 15331183]
Baker MC, Liu Y, Lu R, Lin J, Melehani J, Robinson WH. Incidence of Interstitial Lung Disease in Patients With Rheumatoid Arthritis Treated With Biologic and Targeted Synthetic Disease-Modifying Antirheumatic Drugs. JAMA network open. 2023 Mar 1:6(3):e233640. doi: 10.1001/jamanetworkopen.2023.3640. Epub 2023 Mar 1 [PubMed PMID: 36939701]
Kumar A, Cherian SV, Vassallo R, Yi ES, Ryu JH. Current Concepts in Pathogenesis, Diagnosis, and Management of Smoking-Related Interstitial Lung Diseases. Chest. 2018 Aug:154(2):394-408. doi: 10.1016/j.chest.2017.11.023. Epub 2017 Dec 5 [PubMed PMID: 29222007]
Tillotson CV, Reynolds SB, Patel BC. Langerhans Cell Histiocytosis. StatPearls. 2026 Jan:(): [PubMed PMID: 28613635]
Root MZ, Lee JS. Cryptogenic Organizing Pneumonia. Seminars in respiratory and critical care medicine. 2025 Oct 23:():. doi: 10.1055/a-2703-4537. Epub 2025 Oct 23 [PubMed PMID: 40967604]
Hino T, Lee KS, Yoo H, Han J, Franks TJ, Hatabu H. Interstitial lung abnormality (ILA) and nonspecific interstitial pneumonia (NSIP). European journal of radiology open. 2021:8():100336. doi: 10.1016/j.ejro.2021.100336. Epub 2021 Mar 16 [PubMed PMID: 33796637]
Spagnolo P, Rossi G, Trisolini R, Sverzellati N, Baughman RP, Wells AU. Pulmonary sarcoidosis. The Lancet. Respiratory medicine. 2018 May:6(5):389-402. doi: 10.1016/S2213-2600(18)30064-X. Epub 2018 Apr 3 [PubMed PMID: 29625772]
Hu ZW, Gao L, Yu Q, Jin Z, Liu JH, Lian YY, Que CL. Use of 6-minute walk test for assessing severity of interstitial lung disease: an observational study. Sarcoidosis, vasculitis, and diffuse lung diseases : official journal of WASOG. 2023 Jun 29:40(2):e2023013. doi: 10.36141/svdld.v40i2.13991. Epub 2023 Jun 29 [PubMed PMID: 37382072]
Level 2 (mid-level) evidencePodolanczuk AJ, Hunninghake GM, Wilson KC, Khor YH, Kheir F, Pang B, Adegunsoye A, Cararie G, Corte TJ, Flanagan J, Gudmundsson G, Hariri LP, Hatabu H, Humphries SM, Kaul B, Kim JS, Konigshoff M, Kropski JA, Lee JS, Luo F, Lynch DA, Martinez FJ, Montesi SB, Moodley Y, Oldham JM, Piciucchi S, Putman RK, Richeldi L, Rosas IO, Salisbury ML, Salvatore MM, Selman M, Seo JB, Song JW, Thomson CC, Vivero M, Wain LV, Wijsenbeek M, Schwartz DA, Ryerson CJ. Approach to the Evaluation and Management of Interstitial Lung Abnormalities: An Official American Thoracic Society Clinical Statement. American journal of respiratory and critical care medicine. 2025 Jul:211(7):1132-1155. doi: 10.1164/rccm.202505-1054ST. Epub [PubMed PMID: 40387336]
Kondoh Y, Taniguchi H, Yokoi T, Nishiyama O, Ohishi T, Kato T, Suzuki K, Suzuki R. Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. The European respiratory journal. 2005 Mar:25(3):528-33 [PubMed PMID: 15738299]
Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. European respiratory review : an official journal of the European Respiratory Society. 2019 Mar 31:28(151):. doi: 10.1183/16000617.0100-2018. Epub 2019 Feb 27 [PubMed PMID: 30814139]
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme-Beaty E, Rosenstock B, Quaresma M, Haeufel T, Goeldner RG, Schlenker-Herceg R, Brown KK, INBUILD Trial Investigators. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. The New England journal of medicine. 2019 Oct 31:381(18):1718-1727. doi: 10.1056/NEJMoa1908681. Epub 2019 Sep 29 [PubMed PMID: 31566307]
Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M, Clerisme-Beaty E, Stowasser S, Tetzlaff K, Kuwana M, Maher TM, SENSCIS Trial Investigators. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. The New England journal of medicine. 2019 Jun 27:380(26):2518-2528. doi: 10.1056/NEJMoa1903076. Epub 2019 May 20 [PubMed PMID: 31112379]
Wu S, Xu W, Bei Z, Wu J, Zhang M. Treatment of Systemic Sclerosis-Associated Interstitial Lung Disease: A Systematic Review and Network Meta-Analysis. Archives of rheumatology. 2025 Sep 1:40(3):395-406. doi: 10.5152/ArchRheumatol.2025.25013. Epub [PubMed PMID: 40977300]
Level 1 (high-level) evidenceKhanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, Allanore Y, Matucci-Cerinic M, Distler O, Shima Y, van Laar JM, Spotswood H, Wagner B, Siegel J, Jahreis A, Denton CP, focuSSced investigators. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. Respiratory medicine. 2020 Oct:8(10):963-974. doi: 10.1016/S2213-2600(20)30318-0. Epub 2020 Aug 28 [PubMed PMID: 32866440]
Level 1 (high-level) evidenceCassady SJ, Ramani GV, Wu B, Morland K, Burger CD. Real-World Comparison of Patients With PH-ILD Initiating Inhaled Treprostinil Versus Patients Who Remain Untreated. Pulmonary circulation. 2025 Oct:15(4):e70203. doi: 10.1002/pul2.70203. Epub 2025 Nov 10 [PubMed PMID: 41221470]
Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, Levvey BJ, Lund LH, Meiser B, Rossano JW, Stehlik J. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report--2015; Focus Theme: Early Graft Failure. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015 Oct:34(10):1264-77. doi: 10.1016/j.healun.2015.08.014. Epub 2015 Sep 3 [PubMed PMID: 26454740]
Richeldi L, Azuma A, Cottin V, Kreuter M, Maher TM, Martinez FJ, Oldham JM, Valenzuela C, Clerisme-Beaty E, Gordat M, Wachtlin D, Liu Y, Schlecker C, Stowasser S, Zoz DF, Wijsenbeek MS, FIBRONEER-IPF Trial Investigators. Nerandomilast in Patients with Idiopathic Pulmonary Fibrosis. The New England journal of medicine. 2025 Jun 12:392(22):2193-2202. doi: 10.1056/NEJMoa2414108. Epub 2025 May 18 [PubMed PMID: 40387033]
Fabyan KD, Chandel A, King CS. Pulmonary Hypertension in Interstitial Lung Disease: Management Options to Move Beyond Supportive Care. Current pulmonology reports. 2023 May 22:():1-8. doi: 10.1007/s13665-023-00311-2. Epub 2023 May 22 [PubMed PMID: 37362782]
Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ, King TE Jr, Collard HR. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2011 Dec 15:184(12):1390-4. doi: 10.1164/rccm.201101-0138OC. Epub 2011 Jun 23 [PubMed PMID: 21700909]
Sun JW, Hsu HC, Wu JS, Huang TW, Tsai YS, Cheng WH, Lin LY. Sleep apnea in interstitial lung disease: A systematic review and meta-analysis of prevalence, severity, and risk factors. Sleep medicine. 2025 Dec:136():106768. doi: 10.1016/j.sleep.2025.106768. Epub 2025 Aug 29 [PubMed PMID: 40976082]
Level 1 (high-level) evidenceGreen H, Paul M, Vidal L, Leibovici L. Prophylaxis of Pneumocystis pneumonia in immunocompromised non-HIV-infected patients: systematic review and meta-analysis of randomized controlled trials. Mayo Clinic proceedings. 2007 Sep:82(9):1052-9 [PubMed PMID: 17803871]
Level 1 (high-level) evidenceSeymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. American journal of respiratory and critical care medicine. 2002 Jul 15:166(2):215-35 [PubMed PMID: 12119235]
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, Lee JS, Leslie KO, Lynch DA, Matteson EL, Mosca M, Noth I, Richeldi L, Strek ME, Swigris JJ, Wells AU, West SG, Collard HR, Cottin V, “ERS/ATS Task Force on Undifferentiated Forms of CTD-ILD”. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. The European respiratory journal. 2015 Oct:46(4):976-87. doi: 10.1183/13993003.00150-2015. Epub 2015 Jul 9 [PubMed PMID: 26160873]
Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE Jr, Lancaster L, Sahn SA, Szwarcberg J, Valeyre D, du Bois RM, CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet (London, England). 2011 May 21:377(9779):1760-9. doi: 10.1016/S0140-6736(11)60405-4. Epub 2011 May 13 [PubMed PMID: 21571362]
Level 1 (high-level) evidenceRicheldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR, INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. The New England journal of medicine. 2014 May 29:370(22):2071-82. doi: 10.1056/NEJMoa1402584. Epub 2014 May 18 [PubMed PMID: 24836310]
Level 1 (high-level) evidenceWells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, Moua T, Crestani B, Wuyts WA, Stowasser S, Quaresma M, Goeldner RG, Schlenker-Herceg R, Kolb M, INBUILD trial investigators. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. The Lancet. Respiratory medicine. 2020 May:8(5):453-460. doi: 10.1016/S2213-2600(20)30036-9. Epub 2020 Mar 5 [PubMed PMID: 32145830]
Level 1 (high-level) evidenceTashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, Goldin J, Arriola E, Volkmann ER, Kafaja S, Silver R, Steen V, Strange C, Wise R, Wigley F, Mayes M, Riley DJ, Hussain S, Assassi S, Hsu VM, Patel B, Phillips K, Martinez F, Golden J, Connolly MK, Varga J, Dematte J, Hinchcliff ME, Fischer A, Swigris J, Meehan R, Theodore A, Simms R, Volkov S, Schraufnagel DE, Scholand MB, Frech T, Molitor JA, Highland K, Read CA, Fritzler MJ, Kim GHJ, Tseng CH, Elashoff RM, Sclerodema Lung Study II Investigators. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. The Lancet. Respiratory medicine. 2016 Sep:4(9):708-719. doi: 10.1016/S2213-2600(16)30152-7. Epub 2016 Jul 25 [PubMed PMID: 27469583]
Level 1 (high-level) evidenceBehr J, Demedts M, Buhl R, Costabel U, Dekhuijzen RP, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Lankhorst I, Sardina M, Boissard G, IFIGENIA study group. Lung function in idiopathic pulmonary fibrosis--extended analyses of the IFIGENIA trial. Respiratory research. 2009 Oct 27:10(1):101. doi: 10.1186/1465-9921-10-101. Epub 2009 Oct 27 [PubMed PMID: 19860915]
Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. The New England journal of medicine. 2014 May 29:370(22):2093-101. doi: 10.1056/NEJMoa1401739. Epub 2014 May 18 [PubMed PMID: 24836309]
Level 1 (high-level) evidenceKing TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW, ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. The New England journal of medicine. 2014 May 29:370(22):2083-92. doi: 10.1056/NEJMoa1402582. Epub 2014 May 18 [PubMed PMID: 24836312]
du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, King TE Jr, Lancaster L, Noble PW, Sahn SA, Thomeer M, Valeyre D, Wells AU. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. American journal of respiratory and critical care medicine. 2011 Dec 15:184(12):1382-9. doi: 10.1164/rccm.201105-0840OC. Epub 2011 Sep 22 [PubMed PMID: 21940789]
Nathan SD, Shlobin OA, Weir N, Ahmad S, Kaldjob JM, Battle E, Sheridan MJ, du Bois RM. Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest. 2011 Jul:140(1):221-229. doi: 10.1378/chest.10-2572. Epub [PubMed PMID: 21729893]
Sprunger DB, Olson AL, Huie TJ, Fernandez-Perez ER, Fischer A, Solomon JJ, Brown KK, Swigris JJ. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease. The European respiratory journal. 2012 Jan:39(1):125-32. doi: 10.1183/09031936.00041411. Epub 2011 Jul 7 [PubMed PMID: 21737559]
Lee YJ, Choi SM, Lee YJ, Cho YJ, Yoon HI, Lee JH, Lee CT, Park JS. Clinical impact of depression and anxiety in patients with idiopathic pulmonary fibrosis. PloS one. 2017:12(9):e0184300. doi: 10.1371/journal.pone.0184300. Epub 2017 Sep 11 [PubMed PMID: 28892504]
Dowman L, Hill CJ, May A, Holland AE. Pulmonary rehabilitation for interstitial lung disease. The Cochrane database of systematic reviews. 2021 Feb 1:2(2):CD006322. doi: 10.1002/14651858.CD006322.pub4. Epub 2021 Feb 1 [PubMed PMID: 34559419]
Level 1 (high-level) evidenceWalsh SL, Calandriello L, Sverzellati N, Wells AU, Hansell DM, UIP Observer Consort. Interobserver agreement for the ATS/ERS/JRS/ALAT criteria for a UIP pattern on CT. Thorax. 2016 Jan:71(1):45-51. doi: 10.1136/thoraxjnl-2015-207252. Epub 2015 Nov 19 [PubMed PMID: 26585524]
Akiyama N, Fujisawa T, Morita T, Koyauchi T, Matsuda Y, Mori M, Miyashita M, Tachikawa R, Tomii K, Tomioka H, Hagimoto S, Kondoh Y, Inoue Y, Suda T. End-of-life care for idiopathic pulmonary fibrosis patients with acute exacerbation. Respiratory research. 2022 Oct 29:23(1):294. doi: 10.1186/s12931-022-02204-5. Epub 2022 Oct 29 [PubMed PMID: 36309741]
Sciriha A, Lungaro-Mifsud S, Fsadni P, Scerri J, Montefort S. Pulmonary Rehabilitation in patients with Interstitial Lung Disease: The effects of a 12-week programme. Respiratory medicine. 2019 Jan:146():49-56. doi: 10.1016/j.rmed.2018.11.007. Epub 2018 Nov 23 [PubMed PMID: 30665518]