Introduction
Diffuse alveolar hemorrhage (DAH) represents a severe and potentially life-threatening clinical syndrome characterized by the accumulation of blood within the alveolar spaces. Clinically, DAH classically presents with the triad of hemoptysis, progressive iron-deficiency anemia, and diffuse pulmonary infiltrates, although these findings may appear inconsistently, particularly in children. DAH does not constitute a single disease entity; instead, it serves as a shared clinical manifestation of a heterogeneous group of disorders affecting the alveolar-capillary interface. The primary challenge in evaluating DAH is identifying the underlying etiology through a structured, comprehensive diagnostic approach that systematically excludes infectious, immune-mediated, cardiovascular, and structural causes. A diagnosis of idiopathic pulmonary hemosiderosis should only be considered after rigorous exclusion of alternative etiologies.
Idiopathic pulmonary hemosiderosis represents a rare disorder characterized by recurrent episodes of diffuse alveolar hemorrhage that progressively lead to respiratory complications and permanent lung damage. Repeated intra-alveolar bleeding drives chronic pulmonary injury, resulting in substantial morbidity and mortality. Although the precise etiology remains incompletely understood, current evidence suggests autoimmune-mediated injury to the alveolar capillaries, leading to recurrent hemorrhage within the alveolar spaces. Accumulating data increasingly support a fundamentally immune-mediated mechanism, prompting some investigators to propose the term “immune-mediated pulmonary hemosiderosis” to more accurately reflect the likely underlying pathobiology.[1] Idiopathic pulmonary hemosiderosis now falls within the broader classification of children’s interstitial lung disease (chILD), a framework that has improved diagnostic standardization and facilitated collaborative research efforts.[2][3]
Diagnosis of idiopathic pulmonary hemosiderosis requires exclusion of other causes of primary and secondary pulmonary hemosiderosis through thorough clinical evaluation and systematic diagnostic investigation. Clinicians should maintain a high index of suspicion for recurrent occult alveolar hemorrhage in patients with unexplained iron-deficiency anemia, even in the absence of hemoptysis or overt respiratory symptoms, particularly among pediatric patients.[4] Early recognition and timely diagnosis remain essential for reducing the substantial morbidity and mortality associated with this condition.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of idiopathic pulmonary hemosiderosis remains incompletely understood and is likely multifactorial. Current evidence supports immune dysregulation as the predominant underlying mechanism, although a definitive unifying pathway has not been established (see Image. Immune-Mediated Pulmonary Hemosiderosis).[1][5] Idiopathic pulmonary hemosiderosis is therefore best conceptualized as a syndrome that may arise from heterogeneous triggers acting on a susceptible host.
Proposed contributing factors include environmental exposures, developmental vulnerability, and genetic or immune predisposition. Early epidemiologic studies suggested associations with toxic environmental exposures, including agricultural insecticides and indoor molds, eg, Stachybotrys chartarum. Notably, a cluster of infantile pulmonary hemorrhage cases in Cleveland in the 1990s was initially linked to Stachybotrys exposure; however, subsequent investigations failed to confirm a causal relationship, and this association remains unproven. Prematurity has also been reported more frequently among affected infants, although whether this reflects increased susceptibility or ascertainment bias is unclear.
The strongest and most reproducible evidence supports an immune-mediated basis. Idiopathic pulmonary hemosiderosis has been reported in association with multiple autoimmune conditions, including rheumatoid arthritis, autoimmune hemolytic anemia, thyroid disease, and celiac disease.[6] In some patients, idiopathic pulmonary hemosiderosis may precede the later development of systemic autoimmune disease, including antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, suggesting that idiopathic pulmonary hemosiderosis may represent an early or form fruste manifestation of immune-mediated pulmonary capillaritis in a subset of cases.[3]
Lane–Hamilton Syndrome
The coexistence of idiopathic pulmonary hemosiderosis and celiac disease, termed Lane–Hamilton syndrome, represents the most clinically significant and potentially modifiable association. The reported prevalence of celiac disease among patients with idiopathic pulmonary hemosiderosis varies widely across cohorts but is consistently higher than in the general population.[7][8] Importantly, gastrointestinal manifestations are frequently absent, and pulmonary symptoms may be the sole presentation; in one systematic review, only approximately 20% of patients had significant gastrointestinal symptoms at diagnosis.[8]
Gluten exposure is thought to trigger or exacerbate alveolar hemorrhage through immune-mediated mechanisms, and hemoptysis may be the only presenting symptom.[9] Notably, initiation of a strict gluten-free diet has been associated with clinical improvement and, in some cases, remission of pulmonary symptoms. Given these observations, routine serologic screening for celiac disease is recommended in all patients with suspected or confirmed idiopathic pulmonary hemosiderosis, regardless of gastrointestinal symptoms.
Other Associated Conditions and Diagnostic Considerations
Other conditions associated with idiopathic pulmonary hemosiderosis include:
- Down syndrome: Approximately 20% of pediatric idiopathic pulmonary hemosiderosis patients in the French RespiRare cohort have Down syndrome, with autoimmune markers present in up to 75% of these patients.[2] This likely reflects an underlying predisposition to immune dysregulation rather than a direct causal relationship.
- Kabuki syndrome (KMT2D/KDM6A mutations): Rarely reported association; limited data suggest a potential immune-mediated mechanism and responsiveness to immunosuppressive therapy, including methylprednisolone pulse therapy after failure of standard prednisolone.[10]
- Heiner syndrome: A non-IgE–mediated hypersensitivity to cow’s milk protein that can present with pulmonary hemosiderosis in infants and young children. This entity must be excluded before diagnosing idiopathic pulmonary hemosiderosis, as it represents a distinct and potentially reversible cause of diffuse alveolar hemorrhage.
- Evolution to systemic vasculitis: A subset of patients initially diagnosed with idiopathic pulmonary hemosiderosis later develop ANCA-associated vasculitis, reinforcing the concept that idiopathic pulmonary hemosiderosis may, in some cases, represent an early or incomplete autoimmune phenotype.[3]
Epidemiology
Due to the rare nature of idiopathic pulmonary hemosiderosis, the incidence and prevalence of the disease are relatively unknown. Many patients previously reported to have idiopathic pulmonary hemosiderosis were likely misclassified and would now fall under the broader category of DAH. A Swedish study from 1984 estimated an incidence of 0.24 cases per million children per year, using data collected from 1950 to 1979.[11] A retrospective study from Japan estimated approximately 1.23 cases per million per year.[12]
Approximately 80% of cases occur in children, most of whom are diagnosed in the first decade of life. However, this estimate is based on aggregated case series rather than population-based cohorts.[13] The largest multicenter international study of pediatric DAH to date, including 124 patients from 26 centers across 15 countries, identified idiopathic pulmonary hemosiderosis as the most frequent single diagnosis within pediatric DAH cohorts.[4] A parallel European multicenter analysis found that approximately 26% of all pediatric DAH patients had no identifiable etiology.[3]
Adult-onset idiopathic pulmonary hemosiderosis accounts for approximately 20% of cases; however, an unknown fraction may represent undiagnosed childhood-onset idiopathic pulmonary hemosiderosis. The gender distribution appears balanced in childhood-onset idiopathic pulmonary hemosiderosis, whereas adult-onset disease shows a male predominance. Adult data from a systematic review demonstrated a male predominance of 64.3% with a median age at diagnosis of 27 years.[5]
Additionally, familial clustering has been noted in several reports, suggesting a genetic component.[14][15][16] Diagnostic delay remains a significant problem: in a contemporary Chinese pediatric cohort, the mean time from symptom onset to diagnosis was 5 months (range 0.25–36 months), and the majority of patients were initially misdiagnosed, most commonly as iron-deficiency anemia or gastrointestinal bleeding.[17]
Pathophysiology
Diffuse alveolar hemorrhage in idiopathic pulmonary hemosiderosis is characterized by recurrent leakage of erythrocytes into the alveolar spaces, followed by phagocytosis by alveolar macrophages. These macrophages degrade erythrocytes, leading to the accumulation of iron derived from heme metabolism. Intracellular iron is initially stored as ferritin and subsequently converted into hemosiderin within lysosomes. Over time, repeated hemorrhagic episodes result in progressive accumulation of hemosiderin-laden macrophages.
Although iron is sequestered within macrophages, systemic iron deficiency in idiopathic pulmonary hemosiderosis primarily reflects chronic, recurrent alveolar blood loss coupled with inefficient recycling of iron from pulmonary stores. Excess intracellular iron also contributes to lung injury. Iron can catalyze the formation of reactive oxygen species through redox cycling, leading to oxidative stress, epithelial and endothelial injury, and activation of pro-fibrotic pathways. Recurrent hemorrhage and oxidative injury together are thought to drive progressive alveolar damage and, in some cases, interstitial fibrosis.
Emerging Pathogenetic Mechanisms
A proposed mechanism suggests that diffuse alveolar hemorrhage in idiopathic pulmonary hemosiderosis may be mediated by bioactive proteins, including histamine, eosinophilic cationic protein (ECP), and vascular endothelial growth factor (VEGF), which may increase permeability of the alveolar-capillary barrier and facilitate erythrocyte extravasation.[18] This remains a hypothesis based on limited observational and review-level data rather than on established mechanistic evidence. This hypothesis may help explain the absence of immune complex deposition on immunofluorescence, a feature that distinguishes idiopathic pulmonary hemosiderosis from classic immune complex–mediated diffuse alveolar hemorrhage. However, current evidence remains limited, and this mechanism requires further validation.
Alveolar macrophages may also contribute to ongoing injury beyond iron accumulation by releasing proinflammatory mediators, thereby perpetuating a cycle of inflammation and hemorrhage.
Immune Dysregulation
Autoantibodies are detectable in approximately 26.4% of pediatric patients with idiopathic pulmonary hemosiderosis, with antinuclear antibodies (ANA) and antineutrophil cytoplasmic antibodies (ANCA) most commonly detected.[19] The prevalence appears lower in adults (~13.2%).[20] These data are derived from heterogeneous studies with variable antibody panels, titers, and timing of testing (often after treatment initiation), which limits causal interpretation. These findings support a role for immune dysregulation, even in the absence of overt systemic autoimmune disease at presentation, and raise the possibility that idiopathic pulmonary hemosiderosis may represent an early or incomplete autoimmune phenotype in a subset of patients.
Classification of Pulmonary Hemosiderosis
Pulmonary hemosiderosis is traditionally categorized into 3 groups based on underlying pathophysiology:
- Group 1: Antiglomerular basement membrane (anti-GBM) disease
- Characterized by circulating anti-GBM antibodies leading to small-vessel vasculitis with linear deposition of immunoglobulin G along basement membranes. This includes Goodpasture syndrome, in which pulmonary hemorrhage is often accompanied by rapidly progressive glomerulonephritis.
- Approximately 40% to 60% of patients develop alveolar hemorrhage. Diagnosis is typically established by a kidney biopsy demonstrating linear IgG deposition on immunofluorescence rather than a lung biopsy.[21][22]
- Group 2: Immune-mediated diffuse alveolar hemorrhage (capillaritis-predominant)
- Caused by inflammatory injury to the alveolar capillary bed, often mediated by small-vessel vasculitis rather than classic immune complex deposition alone. This results in capillaritis, disruption of the alveolar-capillary barrier, and hemorrhage. Recurrent injury can lead to fibrosis.
- Associated conditions include systemic lupus erythematosus, IgA vasculitis (Henoch–Schönlein purpura), ANCA-associated vasculitides (eg, granulomatosis with polyangiitis, microscopic polyangiitis), and mixed connective tissue disease.[23][24][25]
- Group 3: Idiopathic pulmonary hemosiderosis
- Defined by recurrent alveolar hemorrhage in the absence of identifiable immune-mediated, infectious, cardiovascular, or structural causes.
- Idiopathic pulmonary hemosiderosis remains a diagnosis of exclusion. A descriptive subclassification has been proposed, grouping patients based on features, eg, autoimmune markers, eosinophilia, renal involvement, or multiorgan disease, which may have implications for prognosis and management.[3]
Histopathology
Histopathologic findings in idiopathic pulmonary hemosiderosis vary according to the stage and chronicity of disease progression. Early or acute phases demonstrate intra-alveolar hemorrhage with accumulation of erythrocytes and numerous hemosiderin-laden macrophages within the alveolar spaces. Hemosiderin deposition may also extend into the interstitium. Recurrent or chronic hemorrhagic episodes promote progressive interstitial thickening and fibrosis, reflecting persistent cycles of pulmonary injury and repair.
The hallmark histopathologic characteristic of idiopathic pulmonary hemosiderosis involves a pattern of “bland” alveolar hemorrhage rather than a distinct pathognomonic lesion. Pulmonary capillaritis remains absent, with no neutrophilic infiltration, fibrinoid necrosis of the alveolar septa, or leukocytoclasia identified on histologic examination. Immunofluorescence studies also demonstrate negative findings for immune complex or immunoglobulin deposition.
These negative histopathologic findings are of substantial diagnostic importance because they help distinguish idiopathic pulmonary hemosiderosis from alternative causes of diffuse alveolar hemorrhage, including antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, anti-GBM disease, and connective tissue disease–associated capillaritis. Interpretation nevertheless requires caution, as sampling error may occur and inflammatory features may occasionally be obscured in early-stage or previously treated disease.
History and Physical
The clinical presentation of idiopathic pulmonary hemosiderosis varies from an acute onset illness with hemoptysis and dyspnea to a chronic cough and dyspnea to repetitive hemoptysis with fatigue, anemia, and slowly progressive dyspnea. In some cases, asymptomatic anemia is the only finding present. Contemporary multicenter data demonstrate that anemia is the most prominent presenting feature in children, present in approximately 87% of patients in international pediatric DAH cohorts, while hemoptysis occurs in approximately 42% and is frequently absent in younger children.[4] Higher rates of hemoptysis have been reported in idiopathic pulmonary hemosiderosis-specific cohorts, reflecting heterogeneity across studies.[17] Unexplained iron-deficiency anemia, even without respiratory symptoms, should therefore always prompt evaluation for occult alveolar hemorrhage. In adults, respiratory symptoms tend to be more prominent; however, children present with failure to thrive and anemia.[26]
The classic triad of hemoptysis, iron-deficiency anemia, and diffuse pulmonary infiltrates is present in approximately 79% of adult patients and is frequently incomplete at initial presentation.[5] The mean diagnostic delay in contemporary pediatric cohorts remains approximately 5 months, with the majority of patients initially misdiagnosed.[17] Idiopathic pulmonary hemosiderosis presents in 2 phases. The first, as an acute phase, corresponds with intra-alveolar bleeding episodes associated with cough, dyspnea, hemoptysis, and potentially respiratory failure. The second chronic phase is characterized by the gradual resolution of prior symptoms, with or without treatment.
Patients who present in the acute phase of the disease show a wide variety of signs and symptoms, including but not limited to respiratory failure, cough, hemoptysis, and worsening anemia. However, many patients may present with a normal physical exam. Rapid asphyxiation due to massive pulmonary hemorrhage has also been reported. Generally, patients who present in the chronic phase of the disease demonstrate exam findings of pallor, emaciation, hepatosplenomegaly, failure to thrive, or even a completely normal exam. In patients with fibrosis, bilateral crackles and finger clubbing may also be present.[2][27] In patients with Lane-Hamilton syndrome, hemoptysis may be the only presenting symptom, with no gastrointestinal manifestations.[8][9] Clinicians should therefore ask specifically about prior celiac disease diagnosis and dietary gluten exposure in all patients with suspected idiopathic pulmonary hemosiderosis.
Evaluation
The evaluation of suspected idiopathic pulmonary hemosiderosis should follow a structured diagnostic approach of exclusion within the broader framework of DAH (see Image. Idiopathic Pulmonary Hemosiderosis Evaluation). The figure illustrates the diagnostic workup of idiopathic pulmonary hemosiderosis as a structured exclusion pathway. Laboratory evaluation (autoimmune panel, celiac serology, core labs) and bronchoscopic/imaging assessment are performed in parallel, converging on lung biopsy when clinically seronegative vasculitis cannot be excluded. Histopathologic confirmation of bland alveolar hemorrhage without capillaritis or immune complex deposition establishes the diagnosis. Whole-exome sequencing is reserved for atypical pediatric cases.
Because DAH represents a common manifestation of diverse conditions, including immune-mediated vasculitis, infection, coagulopathy, cardiac disease, and drug toxicity, no single test confirms idiopathic pulmonary hemosiderosis, and the diagnosis is established only after systematic exclusion of alternative etiologies.
Serologic and Laboratory Evaluation
A comprehensive autoimmune panel is essential and should include ANCA (anti-PR3 and anti-MPO), anti-GBM antibodies, antinuclear antibodies (ANA), anti–double-stranded DNA, extractable nuclear antigens, rheumatoid factor, antiphospholipid antibodies, complement levels (C3, C4), and cryoglobulins. Importantly, seronegative disease does not exclude ANCA-associated vasculitis or pulmonary capillaritis, and tissue confirmation may still be required.
Urinalysis is mandatory to assess for pulmonary–renal syndromes. Findings, eg, hematuria, proteinuria, or red cell casts, should prompt evaluation for anti-GBM disease or ANCA-associated vasculitis and consideration of renal biopsy. Screening for celiac disease should be performed in all patients, regardless of gastrointestinal symptoms, using anti–tissue transglutaminase IgA, total IgA, and anti-endomysial antibodies.[7][8][9] In patients with IgA deficiency, IgG-based testing is required.
Basic laboratory evaluation typically reveals iron-deficiency anemia. Coagulation studies (PT/INR, aPTT, fibrinogen, D-dimer) are necessary to exclude bleeding diatheses. Noninflammatory causes of DAH, including coagulopathies, anticoagulant use, thrombocytopenia, and elevated pulmonary venous pressure, must be systematically excluded. Transthoracic echocardiography is recommended to evaluate for cardiac causes. A detailed medication review is essential to identify potential drug-induced DAH.
Bronchoscopic Evaluation
Bronchoalveolar lavage is a key diagnostic tool and typically demonstrates hemosiderin-laden macrophages and erythrocytes. Importantly, the presence of siderophages supports the diagnosis of DAH but is not specific to idiopathic pulmonary hemosiderosis and may be seen in multiple causes of alveolar hemorrhage.[28] The diagnosis of DAH is supported by progressively bloodier return on sequential lavage aliquots. This finding reflects ongoing alveolar bleeding and is a characteristic feature described in DAH diagnostic frameworks.[29][30]
Perls’ Prussian blue staining is preferred for detecting hemosiderin. Hemosiderin-laden macrophages typically appear 48 to 72 hours after an acute hemorrhagic event; therefore, bronchoalveolar lavage performed very early may yield false-negative results.[30] Microbiologic studies from bronchoalveolar lavage should be performed to exclude infectious causes. A false-negative bronchoalveolar lavage may occur if performed during a quiescent phase between hemorrhagic episodes.
Pulmonary Function Testing and Imaging
Pulmonary function tests typically demonstrate a restrictive pattern. The diffusing capacity for carbon monoxide may be elevated during acute hemorrhage and normal or reduced in chronic disease.[12] Chest radiographs during acute episodes show diffuse alveolar infiltrates, often basilar predominant. High-resolution computed tomography (HRCT) is more sensitive and typically demonstrates ground-glass opacities; chronic disease may show interstitial changes, fibrosis, traction bronchiectasis, or honeycombing.[2][4][31]
Lung Biopsy
Lung biopsy plays a central role in confirming idiopathic pulmonary hemosiderosis, particularly in adults, where seronegative vasculitis cannot be reliably excluded without histopathology. Biopsy may be obtained via transbronchial, cryobiopsy, or surgical approaches, with surgical biopsy providing the highest diagnostic yield.
The histopathologic pattern of idiopathic pulmonary hemosiderosis consists of hemosiderin-laden macrophages in the absence of capillaritis, neutrophilic infiltration, or immune complex deposition, thereby confirming a diagnosis of “bland” alveolar hemorrhage.[2] Lung biopsy is particularly indicated when clinical suspicion for capillaritis remains despite negative serologic testing, as histopathology is required to distinguish bland alveolar hemorrhage from inflammatory capillaritis.
Advanced and Adjunctive Evaluation
In atypical or severe pediatric cases, genetic evaluation, eg, whole-exome sequencing, may be considered, particularly when syndromic features or immunodeficiency are suspected.[10] Following diagnosis, patients may be categorized into descriptive subgroups based on associated features (eg, autoimmune markers, eosinophilia, renal or multiorgan involvement), which may have implications for prognosis and management.[3]
Treatment / Management
No established gold-standard therapy currently exists for idiopathic pulmonary hemosiderosis, and management strategies rely primarily on observational cohorts, case series, and expert consensus rather than randomized controlled trials. Treatment plans require individualization based on disease severity and the nature of the presentation, whether acute or chronic.
First-Line Therapy
Systemic corticosteroids remain the cornerstone of therapy in both pediatric and adult populations.[32] Acute disease commonly requires initiation of prednisolone at dosages of 0.5 to 0.75 mg/kg/day, up to a maximum of 60 mg/day, followed by gradual tapering after clinical and radiographic improvement occurs.[2] Severe presentations complicated by respiratory failure often necessitate intravenous methylprednisolone pulse therapy at doses of 500 to 2000 mg/day for 3 to 5 days in adults or 20 mg/kg/day in children, followed by transition to oral corticosteroids with tapering to maintenance therapy.[33][34] (B3)
Supportive measures, including blood transfusion, may become necessary in patients with severe anemia or hemodynamic instability. Maintenance therapy frequently involves low-dose corticosteroids at 10 to 15 mg/day in adults or 0.1 to 0.3 mg/kg/day in children. Gradual discontinuation may be considered after 18 to 24 months without recurrence.[35](B3)
Steroid-Sparing and Second-Line Immunosuppressive Therapy
Steroid-sparing immunosuppressive agents are used in refractory, relapsing, or steroid-dependent disease. Commonly used agents include azathioprine, cyclophosphamide, hydroxychloroquine (HCQ), and mycophenolate mofetil.[35][36][37] HCQ is the most frequently used maintenance agent, with approximately 64% of clinicians reporting use in survey data.[35] Combination therapy with corticosteroids, HCQ, and azathioprine has shown favorable outcomes in retrospective studies.[17] A randomized phase 2 trial of HCQ in pediatric interstitial lung disease (chILD), which included only 2 patients with idiopathic pulmonary hemosiderosis, did not demonstrate a statistically significant benefit over placebo for its primary endpoint; therefore, its findings should be interpreted as limited to feasibility and safety rather than efficacy in idiopathic pulmonary hemosiderosis.[38](A1)
Mycophenolate mofetil has demonstrated efficacy in both refractory idiopathic pulmonary hemosiderosis and Lane–Hamilton syndrome.[8][9] However, evidence specific to idiopathic pulmonary hemosiderosis remains limited, and extrapolation from associated conditions should be made cautiously. Other agents, including methotrexate, 6-mercaptopurine, and inhaled corticosteroids, have been used with variable success.[35] Clinicians should be aware of potential adverse effects and drug interactions, particularly with HCQ, including rare neuropsychiatric toxicity and interactions with CYP3A4 inhibitors, eg, clarithromycin.[39] (B3)
Lane–Hamilton Syndrome
In patients with Lane–Hamilton syndrome, a strict gluten-free diet is essential and may lead to clinical improvement or remission. Immunosuppressive therapy is used similarly to idiopathic pulmonary hemosiderosis but should be combined with dietary management.[7][8][9](B2)
Rescue and Advanced Therapies
Life-threatening hemorrhage may respond to bronchial artery embolization, which has demonstrated high technical and clinical success rates.[40] Extracorporeal membrane oxygenation (ECMO) may provide support for refractory respiratory failure in experienced centers.[41] Emerging and investigational therapies include liposteroid, or liposomal dexamethasone palmitate, a targeted glucocorticoid therapy supported by limited evidence from small series, and mesenchymal stem cell (MSC) therapy, which has shown immunomodulatory effects and remission in small pediatric cohorts.[42] Current evidence for these approaches remains limited to small case series and early-phase investigations, leaving the efficacy and safety of these approaches uncertain in idiopathic pulmonary hemosiderosis.(B2)
Patients with end-stage disease may require lung transplantation. Recurrence within the allograft has been reported, although recurrence does not occur universally, and idiopathic pulmonary hemosiderosis does not represent an absolute contraindication to transplantation.[43]
Differential Diagnosis
Idiopathic pulmonary hemosiderosis is a diagnosis of exclusion within the DAH spectrum. The following categories must be systematically ruled out before idiopathic pulmonary hemosiderosis can be confirmed.
Immune-Mediated and Vasculitic Causes
These are the most critical exclusions, as they require disease-specific therapy and closely mimic idiopathic pulmonary hemosiderosis. ANCA-associated vasculitides, including granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis, are common mimickers. Seronegativity does not exclude the diagnosis; histologic evidence of capillaritis is required. Anti-GBM disease may present with DAH alone or as a pulmonary–renal syndrome and is confirmed by linear IgG deposition on biopsy. Isolated pulmonary capillaritis can also be seronegative and requires biopsy for differentiation from idiopathic pulmonary hemosiderosis. Connective tissue diseases, including systemic lupus erythematosus (SLE), antiphospholipid syndrome, rheumatoid arthritis, Sjögren syndrome, and mixed connective tissue disease (MCTD), should be considered, as should IgA vasculitis, when systemic or renal features are present. COPA syndrome, a rare genetic interferonopathy, should be considered in pediatric patients with refractory or atypical DAH.
Infectious Causes
Infectious DAH typically presents acutely with systemic features but must always be excluded. Relevant pathogens include bacterial organisms (Streptococcus pneumoniae, Staphylococcus aureus, Legionella pneumophila), respiratory viruses (influenza, RSV), opportunistic fungi (Pneumocystis jirovecii, endemic fungi), and mycobacteria (tuberculosis, nontuberculous mycobacteria).
Noninflammatory and Hemodynamic Causes
These produce bland hemorrhage without capillaritis, making them important mimickers of idiopathic pulmonary hemosiderosis. Causes include inherited and acquired coagulopathies (hemophilia, von Willebrand disease, liver disease, vitamin K deficiency), anticoagulant or antiplatelet therapy, and disseminated intravascular coagulation. Cardiac causes, eg, mitral stenosis, left ventricular failure, and elevated pulmonary venous pressure, must be excluded, as should pulmonary veno-occlusive disease and arteriovenous malformations (AVMs). Please see StatPearls' companion resource, "Pulmonary Arteriovenous Malformation," for further information.
Drug-Induced and Toxic Causes
A thorough medication and exposure history is essential. Implicated agents include amiodarone, nitrofurantoin, infliximab, and penicillamine. E-cigarette or vaping-associated lung injury (EVALI) is an important consideration in adolescents. Inhalational injury, pesticides, and environmental toxins should also be evaluated.
Other Causes and Mimickers
DAH may develop in association with diffuse alveolar damage, acute lung injury syndromes, barotrauma, severe infection, and selected malignant or vascular disorders. Careful evaluation of these alternative etiologies remains essential during diagnostic assessment because multiple pulmonary and systemic conditions can mimic the clinical and radiographic manifestations of DAH. In infants and young children, Heiner syndrome, a cow’s milk protein–associated pulmonary hemosiderosis, requires exclusion before establishing a diagnosis of idiopathic pulmonary hemosiderosis.
An important diagnostic distinction involves Lane–Hamilton syndrome, defined by the coexistence of idiopathic pulmonary hemosiderosis and celiac disease. Lane–Hamilton syndrome does not represent a separate differential diagnosis but rather a clinically significant subtype of idiopathic pulmonary hemosiderosis. All patients with confirmed idiopathic pulmonary hemosiderosis should therefore undergo active screening with celiac serology regardless of the presence or absence of gastrointestinal symptoms.
Prognosis
The prognosis of idiopathic pulmonary hemosiderosis is highly variable, ranging from spontaneous remission to progressive, life-threatening disease. Reported disease duration spans from fulminant cases resulting in death within days to chronic courses extending beyond 2 decades, often complicated by cor pulmonale.
Historical series reported a mean survival of 2.5 to 5 years following diagnosis; however, these data largely reflect the pre-immunosuppression era and underestimate current outcomes. More recent studies demonstrate substantially improved survival, with 5-year survival exceeding 80% to 85% in pediatric patients receiving long-term immunosuppressive therapy[2] and 67% to 84% in adult cohorts.[5][4]
Relapse is common, with approximately 59.4% of pediatric patients experiencing at least one recurrence after initial therapy.[17] Identified risk factors for relapse include lower hemoglobin at presentation and failure to achieve remission within the first 6 months of treatment. Notably, the severity of initial presentation does not reliably predict long-term outcomes.[5]
Mortality may occur acutely due to massive alveolar hemorrhage or chronically from progressive respiratory insufficiency and right heart failure. Even among long-term survivors, persistent morbidity is common in pediatric DAH cohorts, including patients with idiopathic pulmonary hemosiderosis H, with ongoing lung function impairment and structural abnormalities on high-resolution computed tomography (HRCT) reported in a substantial proportion of patients.[4]
Complications
Complications of idiopathic pulmonary hemosiderosis reflect both recurrent alveolar hemorrhage and the consequences of chronic lung injury and immunosuppressive therapy. Their severity depends on disease burden and frequency of relapse.
The most common complications are iron-deficiency anemia and progressive pulmonary fibrosis, both resulting from repeated hemorrhagic episodes. In the acute setting, complications range from hypoxemia and respiratory distress to life-threatening events, including massive alveolar hemorrhage leading to airway compromise, hemodynamic instability, and death. Chronic disease is associated with progressive respiratory insufficiency and may lead to right heart failure (cor pulmonale).
Additional complications reported in contemporary cohorts include:
- Pulmonary arterial hypertension: Particularly in patients with recurrent exacerbations.[2]
- Bronchiectasis: Detected on high-resolution computed tomography (HRCT) in patients with repeated hemorrhagic injury.[17]
- Treatment complications: Treatment-related complications, including Cushingoid features, increased infection risk, and vitamin D deficiency, were reported in approximately 21.9% of pediatric patients.[17]
Consultations
An interprofessional team approach can help to manage patients with idiopathic pulmonary hemosiderosis efficiently. A collaboration between the following specialties is recommended:
- Pulmonology
- Nephrology
- Intensive care unit
- Rheumatology
- Thoracic surgery
- Gastroenterology: for LHS evaluation, celiac disease confirmation (including intestinal biopsy), and dietary management coordination.[7]
- Dietitian/Nutritionist: for patients with LHS requiring counseling on strict adherence to a lifelong gluten-free diet.
- Genetics: for atypical presentations or suspected genetic syndrome (eg, Kabuki syndrome).[10]
Deterrence and Patient Education
Patient education in idiopathic pulmonary hemosiderosis focuses on preventing recurrence, optimizing adherence to therapy, and monitoring for complications. For patients with Lane–Hamilton syndrome, a strict lifelong gluten-free diet is essential and may significantly reduce or prevent hemorrhagic episodes, even in the absence of gastrointestinal symptoms.[7][8][9] In most patients, maintenance therapy with systemic corticosteroids and/or steroid-sparing agents is required to reduce relapse risk and may help delay progression to fibrotic lung disease.
Key components of patient and family education include:
- Celiac disease screening and management: All patients should undergo serologic testing; confirmed cases require strict adherence to a gluten-free diet.[7][8][9]
- Infection prevention: Respiratory infections (particularly influenza and respiratory syncytial virus) may precipitate exacerbations. Routine immunizations should be maintained, with adjustments as needed for immunosuppressed patients.
- Long-term surveillance: Given the high relapse rate (~59%) and risk of progressive lung disease and pulmonary hypertension, patients require ongoing follow-up with pulmonary function testing and periodic high-resolution computed tomography (HRCT).[17]
- Medication adherence and monitoring: Education regarding long-term immunosuppressive therapy, including potential adverse effects and the importance of adherence, is essential.
- Psychosocial support: Families should be directed to appropriate resources, including rare disease registries and pediatric interstitial lung disease (chILD) support networks.
Enhancing Healthcare Team Outcomes
Idiopathic pulmonary hemosiderosis is a rare cause of diffuse alveolar hemorrhage characterized by recurrent bleeding into the alveolar spaces, progressive iron-deficiency anemia, and potential development of pulmonary fibrosis and respiratory failure. Increasing evidence supports immune dysregulation as a major pathogenic mechanism, although the disease remains a diagnosis of exclusion after systematic evaluation for vasculitis, infection, coagulopathy, cardiovascular disease, celiac disease, and structural lung disorders. Clinical presentation ranges from hemoptysis and dyspnea to isolated anemia without respiratory symptoms, particularly in children. Diagnostic evaluation includes autoimmune serologies, bronchoalveolar lavage, pulmonary imaging, lung biopsy, and exclusion of secondary causes. Management commonly involves corticosteroids, steroid-sparing immunosuppressive therapy, supportive care, and gluten-free dietary therapy in patients with Lane–Hamilton syndrome.
Interprofessional collaboration improves diagnostic accuracy, continuity of care, and long-term outcomes in patients with idiopathic pulmonary hemosiderosis. Pulmonologists, rheumatologists, hospitalists, primary care clinicians, and advanced practitioners coordinate diagnostic evaluation, immunosuppressive therapy, and longitudinal monitoring for relapse, fibrosis, pulmonary hypertension, and treatment toxicity. Nurses provide patient education, assess adherence, monitor for clinical deterioration, and facilitate communication across care settings. Pharmacists evaluate medication interactions, optimize immunosuppressive regimens, and support safe long-term corticosteroid use. Radiologists and pathologists contribute essential diagnostic interpretation of imaging and histopathology, while gastroenterologists assist in identifying celiac disease and managing Lane–Hamilton syndrome. Shared decision-making with patients and families supports adherence, informed consent, and individualized treatment planning. Referral to specialized centers and participation in collaborative networks improve access to expertise, promote evidence-based care, and advance research in this rare disease population.[3][4]
Media
(Click Image to Enlarge)
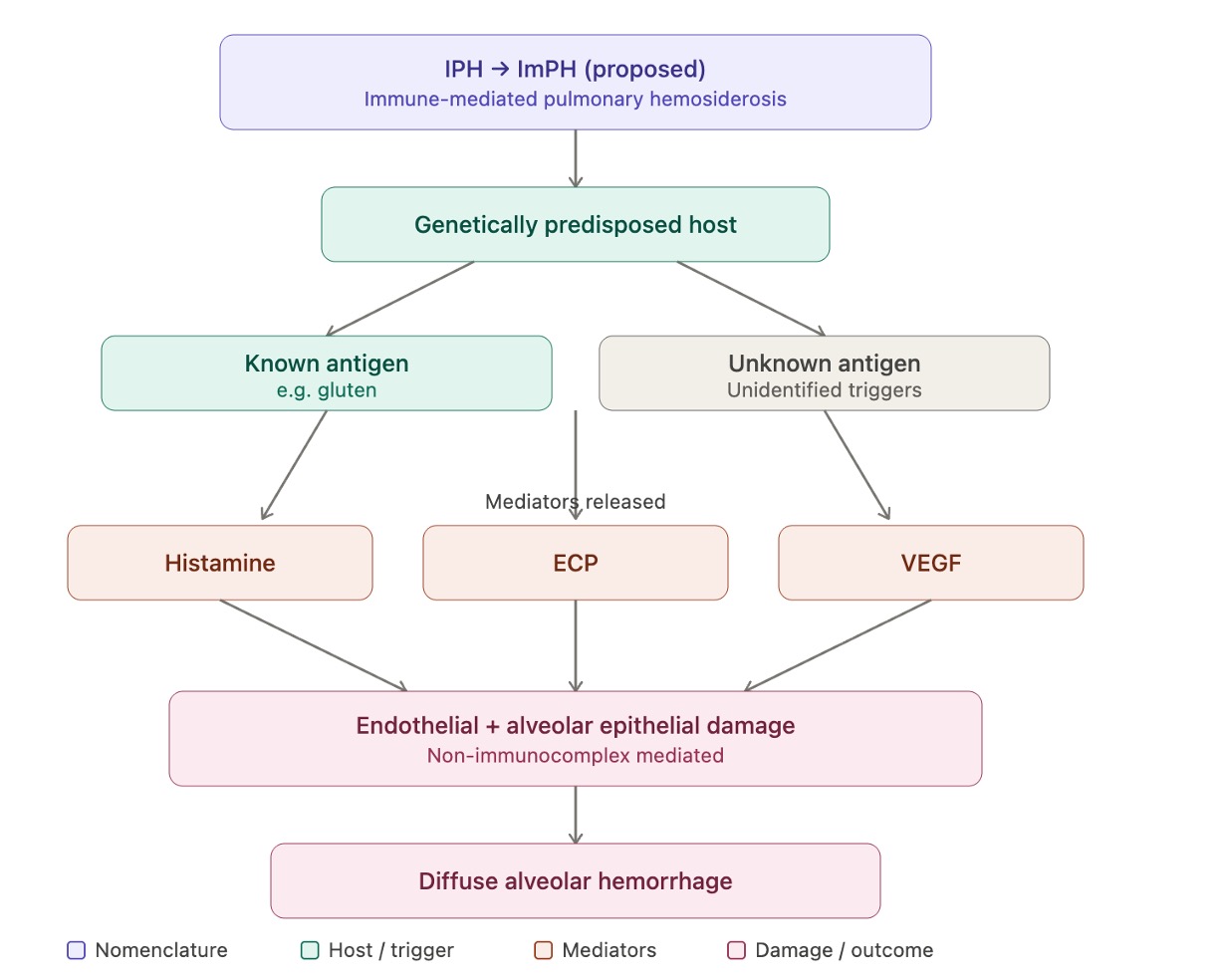
Immune-Mediated Pulmonary Hemosiderosis. Proposed pathogenesis of immune-mediated pulmonary hemosiderosis (ImPH).
Abbreviations: IPH, idiopathic pulmonary hemosiderosis; ImPH, immune-mediated pulmonary hemosiderosis; ECP, eosinophilic cationic protein; VEGF, vascular endothelial growth factor; DAH, diffuse alveolar hemorrhage.
Contributed by A Sankari, MD
(Click Image to Enlarge)
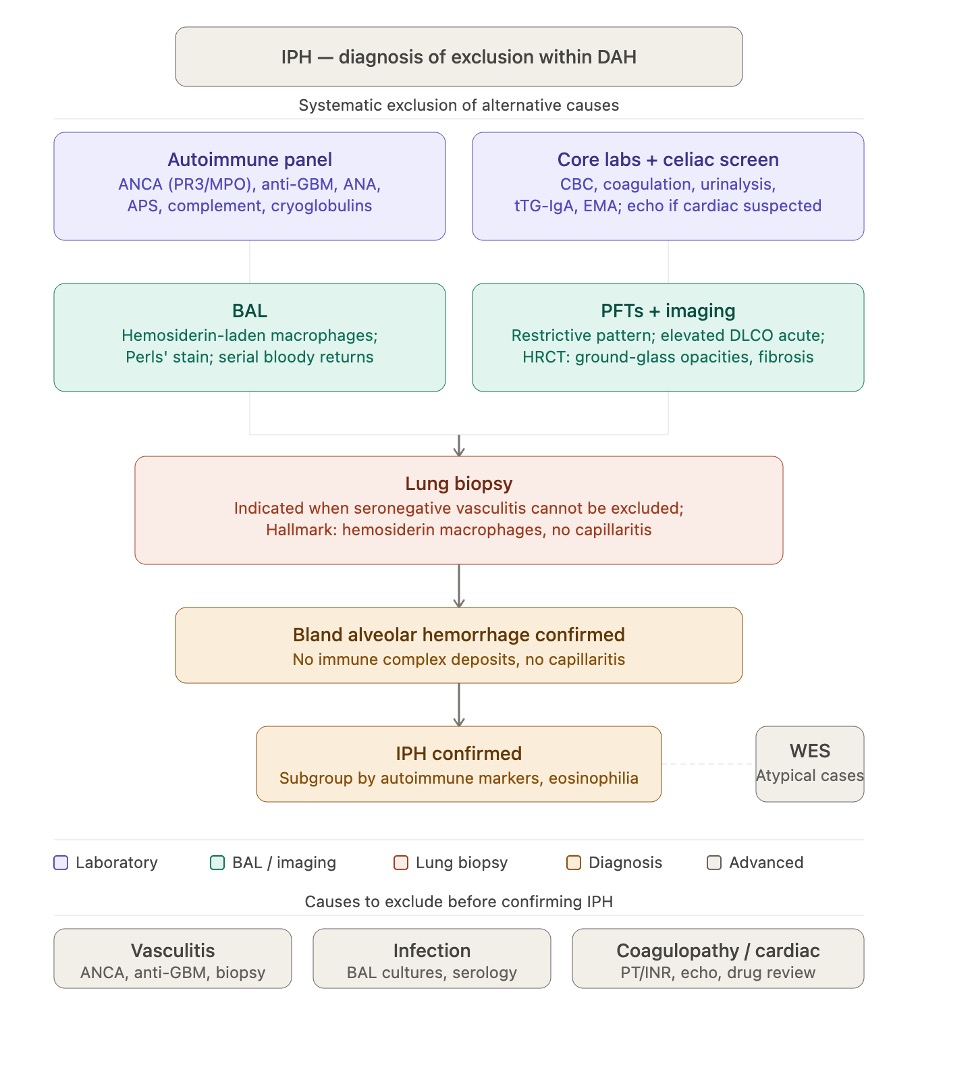
Idiopathic Pulmonary Hemosiderosis Evaluation. The figure illustrates the diagnostic workup of IPH as a structured exclusion pathway.
Abbreviations: ANCA, antineutrophil cytoplasmic antibody; ANA, antinuclear antibody; anti-GBM, anti–glomerular basement membrane; APS, antiphospholipid syndrome; aPTT, activated partial thromboplastin time; BAL, bronchoalveolar lavage; CBC, complete blood count; DAH, diffuse alveolar hemorrhage; DLCO, diffusing capacity for carbon monoxide; EMA, anti-endomysial antibody; GGO, ground-glass opacity; HRCT, high-resolution computed tomography; ICD, immune complex deposition; IPH, idiopathic pulmonary hemosiderosis; PFT, pulmonary function test; PT/INR, prothrombin time/international normalized ratio; TBLB, transbronchial lung biopsy; tTG-IgA, anti–tissue transglutaminase IgA; TTE, transthoracic echocardiography; VATS, video-assisted thoracoscopic surgery; WES, whole-exome sequencing.
Contributed by A Sankari, MD
References
Saha BK. Is It Time to Call Idiopathic Pulmonary Hemosiderosis by the Correct Name: Immune-Mediated Pulmonary Hemosiderosis? The American journal of the medical sciences. 2021 Jun:361(6):809-811. doi: 10.1016/j.amjms.2021.01.006. Epub 2021 Jan 9 [PubMed PMID: 33487400]
Ioachimescu OC, Sieber S, Kotch A. Idiopathic pulmonary haemosiderosis revisited. The European respiratory journal. 2004 Jul:24(1):162-70 [PubMed PMID: 15293620]
Level 3 (low-level) evidenceKnoflach K, Rapp CK, Schwerk N, Carlens J, Wetzke M, Emiralioğlu N, Kiper N, Ring AM, Buchvald F, Manali E, Papiris S, Reu-Hofer S, Kappler M, Schieber A, Seidl E, Gothe F, Robinson PN, Griese M, ChILD EU Collaborators. Diffuse alveolar hemorrhage in children with interstitial lung disease: Determine etiologies! Pediatric pulmonology. 2023 Apr:58(4):1106-1121. doi: 10.1002/ppul.26301. Epub 2023 Jan 13 [PubMed PMID: 36588100]
Ring AM, Schwerk N, Kiper N, Aslan AT, Aurora P, Ayats R, Azevedo I, Bandeira T, Carlens J, Castillo-Corullon S, Cobanoglu N, Elnazir B, Emiralioğlu N, Eyuboglu TS, Fayon M, Gursoy TR, Hogg C, Kötz K, Karadag B, Látalová V, Krenke K, Lange J, Manali ED, Osona B, Papiris S, Proesmann M, Reix P, Roditis L, Rubak S, Rumman N, Snijders D, Stehling F, Weiss L, Yalcın E, Zirek F, Bush A, Clement A, Griese M, Buchvald FF, Nathan N, Nielsen KG. Diffuse alveolar haemorrhage in children: an international multicentre study. ERJ open research. 2023 Mar:9(2):. pii: 00733-2022. doi: 10.1183/23120541.00733-2022. Epub 2023 Apr 24 [PubMed PMID: 37101741]
Saha BK, Bonnier A, Saha S, Saha BN, Shkolnik B. Adult patients with idiopathic pulmonary hemosiderosis: a comprehensive review of the literature. Clinical rheumatology. 2022 Jun:41(6):1627-1640. doi: 10.1007/s10067-022-06104-3. Epub 2022 Feb 18 [PubMed PMID: 35179664]
Saha BK, Chong WH, Milman NT. Differentiation of idiopathic pulmonary hemosiderosis from rheumatologic and autoimmune diseases causing diffuse alveolar hemorrhage: establishing a diagnostic approach. Clinical rheumatology. 2022 Feb:41(2):325-336. doi: 10.1007/s10067-021-05895-1. Epub 2021 Sep 7 [PubMed PMID: 34491458]
Saha BK, Saha S, Bonnier A, Saha BN. Association between idiopathic pulmonary hemosiderosis and celiac disease in pediatric patients: A scoping review of the literature over the past 50 years. Pediatric pulmonology. 2022 May:57(5):1127-1144. doi: 10.1002/ppul.25847. Epub 2022 Feb 11 [PubMed PMID: 35088581]
Level 2 (mid-level) evidenceTryfon S, Papadopoulou E, Psarros G, Agrafiotis M, Saroglou M. Celiac disease and idiopathic pulmonary hemosiderosis: a literature review of the Lane-Hamilton syndrome. Postgraduate medicine. 2022 Nov:134(8):732-742. doi: 10.1080/00325481.2022.2109121. Epub 2022 Aug 8 [PubMed PMID: 35912848]
Grech AK, Yu C. Lane-Hamilton syndrome. Respirology case reports. 2023 Aug:11(8):e01188. doi: 10.1002/rcr2.1188. Epub 2023 Jul 4 [PubMed PMID: 37416498]
Level 3 (low-level) evidenceUejima Y, Yoshida K, Ohashi H. Idiopathic pulmonary hemosiderosis associated with Kabuki syndrome. Immunological medicine. 2024 Dec:47(4):275-277. doi: 10.1080/25785826.2024.2370937. Epub 2024 Jun 25 [PubMed PMID: 38916243]
Kjellman B, Elinder G, Garwicz S, Svan H. Idiopathic pulmonary haemosiderosis in Swedish children. Acta paediatrica Scandinavica. 1984 Sep:73(5):584-8 [PubMed PMID: 6485774]
Ohga S, Takahashi K, Miyazaki S, Kato H, Ueda K. Idiopathic pulmonary haemosiderosis in Japan: 39 possible cases from a survey questionnaire. European journal of pediatrics. 1995 Dec:154(12):994-5 [PubMed PMID: 8801109]
Level 3 (low-level) evidenceMorgan PG, Turner-Warwick M. Pulmonary haemosiderosis and pulmonary haemorrhage. British journal of diseases of the chest. 1981 Jul:75(3):225-42 [PubMed PMID: 7028070]
Beckerman RC, Taussig LM, Pinnas JL. Familial idiopathic pulmonary hemosiderosis. American journal of diseases of children (1960). 1979 Jun:133(6):609-11 [PubMed PMID: 375718]
Level 3 (low-level) evidenceThaell JF, Greipp PR, Stubbs SE, Siegal GP. Idiopathic pulmonary hemosiderosis: two cases in a family. Mayo Clinic proceedings. 1978 Feb:53(2):113-8 [PubMed PMID: 621956]
Level 3 (low-level) evidenceBreckenridge RL Jr, Ross JS. Idiopathic pulmonary hemosiderosis: a report of familial occurrence. Chest. 1979 May:75(5):636-9 [PubMed PMID: 436500]
Level 3 (low-level) evidenceWang L, Li Y, Zhang R, Liu H, Chen L. Clinical features and risk factors for recurrence of idiopathic pulmonary hemosiderosis in children. BMC pulmonary medicine. 2024 Sep 19:24(1):461. doi: 10.1186/s12890-024-03267-4. Epub 2024 Sep 19 [PubMed PMID: 39300433]
Saha BK, Chong WH, Saha S, Aiman A, Bonnier A. Proposed Pathogenesis of Diffuse Alveolar Hemorrhage in Idiopathic Pulmonary Hemosiderosis. Lung. 2022 Apr:200(2):205-215. doi: 10.1007/s00408-022-00523-4. Epub 2022 Mar 10 [PubMed PMID: 35267072]
Saha BK, Bonnier A, Chenna P, Milman NT. Prevalence of autoantibodies in pediatric patients with idiopathic pulmonary hemosiderosis: a scoping review of the literature in the period 1980-2021. Clinical rheumatology. 2022 Apr:41(4):977-990. doi: 10.1007/s10067-021-06029-3. Epub 2022 Jan 24 [PubMed PMID: 35067768]
Level 2 (mid-level) evidenceSaha BK, Bonnier A, Saha S, Saha BN, Milman NT. The Spectrum of Autoantibodies in Adult Patients With Idiopathic Pulmonary Hemosiderosis: A Brief Review of the Literature. Cureus. 2022 Apr:14(4):e24169. doi: 10.7759/cureus.24169. Epub 2022 Apr 15 [PubMed PMID: 35586354]
McAdoo SP, Pusey CD. Anti-Glomerular Basement Membrane Disease. Clinical journal of the American Society of Nephrology : CJASN. 2017 Jul 7:12(7):1162-1172. doi: 10.2215/CJN.01380217. Epub 2017 May 17 [PubMed PMID: 28515156]
Donald KJ, Edwards RL, McEvoy JD. Alveolar capillary basement membrane lesions in Goodpasture's syndrome and idiopathic pulmonary hemosiderosis. The American journal of medicine. 1975 Nov:59(5):642-9 [PubMed PMID: 1106191]
Level 3 (low-level) evidenceTaytard J, Nathan N, de Blic J, Fayon M, Epaud R, Deschildre A, Troussier F, Lubrano M, Chiron R, Reix P, Cros P, Mahloul M, Michon D, Clement A, Corvol H, French RespiRare® group. New insights into pediatric idiopathic pulmonary hemosiderosis: the French RespiRare(®) cohort. Orphanet journal of rare diseases. 2013 Oct 14:8():161. doi: 10.1186/1750-1172-8-161. Epub 2013 Oct 14 [PubMed PMID: 24125570]
Erkoçoğlu M, Civelek E, Kocabaş CN. Unusual presentation: Concurrent IgA deficiency and idiopathic pulmonary hemosiderosis. Pediatric pulmonology. 2016 Oct:51(10):E34-E36. doi: 10.1002/ppul.23445. Epub 2016 Apr 27 [PubMed PMID: 27120139]
Stainer A, Rice A, Devaraj A, Barnett JL, Donovan J, Kokosi M, Nicholson AG, Cairns T, Wells AU, Renzoni EA. Diffuse alveolar haemorrhage associated with subsequent development of ANCA positivity and emphysema in three young adults. BMC pulmonary medicine. 2019 Oct 24:19(1):185. doi: 10.1186/s12890-019-0947-y. Epub 2019 Oct 24 [PubMed PMID: 31651292]
Zhang Y, Luo F, Wang N, Song Y, Tao Y. Clinical characteristics and prognosis of idiopathic pulmonary hemosiderosis in pediatric patients. The Journal of international medical research. 2019 Jan:47(1):293-302. doi: 10.1177/0300060518800652. Epub 2018 Oct 2 [PubMed PMID: 30278795]
Chen XY, Sun JM, Huang XJ. Idiopathic pulmonary hemosiderosis in adults: review of cases reported in the latest 15 years. The clinical respiratory journal. 2017 Nov:11(6):677-681. doi: 10.1111/crj.12440. Epub 2016 Mar 14 [PubMed PMID: 26692115]
Level 3 (low-level) evidenceMeyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, Rottoli P, Saltini C, Selman M, Strange C, Wood B, American Thoracic Society Committee on BAL in Interstitial Lung Disease. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. American journal of respiratory and critical care medicine. 2012 May 1:185(9):1004-14. doi: 10.1164/rccm.201202-0320ST. Epub [PubMed PMID: 22550210]
Level 1 (high-level) evidenceLara AR, Schwarz MI. Diffuse alveolar hemorrhage. Chest. 2010 May:137(5):1164-71. doi: 10.1378/chest.08-2084. Epub [PubMed PMID: 20442117]
Ioachimescu OC, Stoller JK. Diffuse alveolar hemorrhage: diagnosing it and finding the cause. Cleveland Clinic journal of medicine. 2008 Apr:75(4):258, 260, 264-5 passim [PubMed PMID: 18491433]
Bakalli I, Kota L, Sala D, Celaj E, Kola E, Lluka R, Sallabanda S. Idiopathic pulmonary hemosiderosis - a diagnostic challenge. Italian journal of pediatrics. 2014 Apr 4:40():35. doi: 10.1186/1824-7288-40-35. Epub 2014 Apr 4 [PubMed PMID: 24708654]
Level 3 (low-level) evidenceGipsman AI, Grant LMC, Piccione JC, Yehya N, Witmer C, Young LR, Wannes Daou A, Srinivasan A, Phinizy PA. Management of severe acute pulmonary haemorrhage in children. The Lancet. Child & adolescent health. 2025 May:9(5):349-360. doi: 10.1016/S2352-4642(25)00060-4. Epub [PubMed PMID: 40246361]
Deniz O, Ongürü O, Ors F, Gümüş S, Tozkoparan E, Bilgiç H, Ekiz K. Idiopathic pulmonary hemosiderosis in an adult patient responded well to corticosteroid therapy. Tuberkuloz ve toraks. 2007:55(1):77-82 [PubMed PMID: 17401798]
Level 3 (low-level) evidenceLi YT, Guo YX, Cai LM, Pan L, Duan MQ, Yang LF, Sun YY, Tan WP, Chen ZG. Methylprednisolone pulse therapy rescued life-threatening pulmonary hemorrhage due to idiopathic pulmonary hemosiderosis. The American journal of emergency medicine. 2017 Nov:35(11):1786.e3-1786.e7. doi: 10.1016/j.ajem.2017.07.094. Epub 2017 Jul 31 [PubMed PMID: 28784257]
Saha BK, Milman NT. Idiopathic pulmonary hemosiderosis: a review of the treatments used during the past 30 years and future directions. Clinical rheumatology. 2021 Jul:40(7):2547-2557. doi: 10.1007/s10067-020-05507-4. Epub 2020 Nov 12 [PubMed PMID: 33184706]
Level 3 (low-level) evidenceChen CH, Yang HB, Chiang SR, Wang PC. Idiopathic pulmonary hemosiderosis: favorable response to corticosteroids. Journal of the Chinese Medical Association : JCMA. 2008 Aug:71(8):421-4. doi: 10.1016/S1726-4901(08)70094-7. Epub [PubMed PMID: 18772123]
Level 3 (low-level) evidencePal P, De H, Giri PP, Ganguly N, Mandal A. Early Initiation of Steroid-sparing Drugs in Idiopathic Pulmonary Hemosiderosis. Indian pediatrics. 2019 Jan 15:56(1):73-74 [PubMed PMID: 30806370]
Griese M, Kappler M, Stehling F, Schulze J, Baden W, Koerner-Rettberg C, Carlens J, Prenzel F, Nährlich L, Thalmeier A, Sebah D, Kronfeld K, Rock H, Ruckes C, HCQ-study group, Wetzke M, Seidl E, Schwerk N. Randomized controlled phase 2 trial of hydroxychloroquine in childhood interstitial lung disease. Orphanet journal of rare diseases. 2022 Jul 23:17(1):289. doi: 10.1186/s13023-022-02399-2. Epub 2022 Jul 23 [PubMed PMID: 35871071]
Level 1 (high-level) evidenceCengiz DA, Başaran AE, Parlak BB, Şambel IT, Bingöl A. Neuropsychiatric Side Effects of Hydroxychloroquine in a Patient With Idiopathic Pulmonary Hemosiderosis. Journal of paediatrics and child health. 2025 Apr:61(4):639-642. doi: 10.1111/jpc.70001. Epub 2025 Feb 5 [PubMed PMID: 39907062]
Zhao QM, Zhao L, He L, Wu L, Lu Y, Chu C, Wang LB, Niu C, Liu F. Bronchial Artery Embolization in Pediatric Pulmonary Hemorrhage: A Single-Center Experience. Journal of vascular and interventional radiology : JVIR. 2020 Jul:31(7):1103-1109. doi: 10.1016/j.jvir.2019.11.007. Epub 2020 May 23 [PubMed PMID: 32457013]
Matsumoto S, Nakagawa S. Extracorporeal Membrane Oxygenation for Diffuse Alveolar Hemorrhage Caused by Idiopathic Pulmonary Hemosiderosis: A Case Report and a Review of the Literature. Journal of pediatric intensive care. 2019 Sep:8(3):181-186. doi: 10.1055/s-0039-1679904. Epub 2019 Feb 25 [PubMed PMID: 31404435]
Level 3 (low-level) evidenceSaha BK, Milman NT. Liposteroid Therapy for Idiopathic Pulmonary Hemosiderosis: A Scoping Review of the Literature. Prague medical report. 2022:123(2):65-81. doi: 10.14712/23362936.2022.7. Epub [PubMed PMID: 35507939]
Level 2 (mid-level) evidenceSaha BK, Chong WH. Lung transplant to manage end-stage lung disease due to idiopathic pulmonary hemosiderosis: A review of the literature. Respiratory investigation. 2022 Jan:60(1):82-89. doi: 10.1016/j.resinv.2021.06.009. Epub 2021 Jul 24 [PubMed PMID: 34312096]