Introduction
Hypoaldosteronism is a condition marked by insufficient aldosterone production or release from the zona glomerulosa in the adrenal cortex. In cases of pseudohypoaldosteronism, aldosterone levels are often elevated as a compensatory response to peripheral resistance.[1] Aldosterone synthesis depends on the CYP11B2 gene, which is exclusively associated with the zona glomerulosa. This gene encodes the enzyme aldosterone synthase, which catalyzes the final steps in aldosterone biosynthesis—specifically, the conversion of deoxycorticosterone to aldosterone.[2] Research has linked polymorphic variations in the CYP11B2 gene to an increased risk of developing essential hypertension and cardiovascular complications. Implicated polymorphisms include the -344C/T mutation in the gene promoter region and the Lys173Arg mutation in exon 3.[3][4]
The principal regulators of aldosterone synthesis are angiotensin II and potassium. Angiotensin II is part of the renin-angiotensin-aldosterone system (RAAS), the feedback axis that regulates sodium retention, potassium excretion, extracellular fluid volume, and systemic blood pressure. A drop in renal perfusion pressure triggers renin secretion from the macula densa cells of the juxtaglomerular apparatus. Renin cleaves angiotensinogen into angiotensin I. Angiotensin I is converted by angiotensin-converting enzyme into angiotensin II, the most potent stimulus for aldosterone production and release. Serum potassium regulates aldosterone production independently of RAAS. Elevated serum potassium triggers depolarization of zona glomerulosa cells, resulting in the opening of voltage-gated calcium channels and an intracellular calcium influx.[5] The cytosolic calcium binds to calmodulin protein, forming a complex that phosphorylates CYP11B2 transcription factors and upregulates gene transcription. Calcium also promotes the expression of the steroidogenic acute regulatory (StAR) protein. The StAR protein facilitates the transfer of cholesterol into the inner mitochondrial membrane, a rate-limiting step in aldosterone production.[6] Other regulators of aldosterone include adrenocorticotropic hormone and various neuropeptides, although these have a transient and less potent role.[7]
The primary site of action for aldosterone is the mineralocorticoid receptor, an intracellular nuclear receptor located within the principal cells of the renal distal convoluted tubule, connecting tubule, and collecting duct. The density of mineralocorticoid receptors is highest in the distal nephron of the kidney; however, they are also present to a lesser extent in the distal colon, sweat glands, salivary glands, airways, eyes, and in nonepithelial cardiovascular and central nervous tissues.[8] The binding of aldosterone to the mineralocorticoid receptor forms an activated complex that translocates to the nucleus and acts as a transcription factor. This aldosterone-mineralocorticoid receptor complex upregulates the transcription of genes that encode the epithelial sodium channels and the sodium-potassium pump, promoting sodium reabsorption, potassium excretion, and expansion of extracellular fluid.
Both cortisol and aldosterone demonstrate equal affinity for the mineralocorticoid receptor. Physiological serum cortisol concentrations are comparatively higher than aldosterone, partly due to factors, eg, plasma protein binding, half-life duration, and secretion patterns. The enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), primarily located in the renal tubules, converts the active form of cortisol (which can bind to the mineralocorticoid receptor) into the inactive form of cortisone (which cannot bind to the mineralocorticoid receptor). This serves as a regulatory mechanism that prevents excessive stimulation of the mineralocorticoid receptor by cortisol.[9]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Isolated Aldosteron Defects
Isolated aldosterone defects can present as abnormalities in synthesis, release, or end-organ response. Each mechanism reflects a distinct underlying pathophysiology with unique congenital, acquired, or medication-induced contributors. Recognition of these categories supports accurate diagnosis and targeted treatment strategies.
Defective synthesis
Defective synthesis, known as hyperreninemic hypoaldosteronism, occurs when aldosterone production fails despite intact renin secretion. Congenital causes include aldosterone synthase deficiency types I and II (see Image. Aldosterone Synthase Enzyme). Acquired causes involve immaturity of aldosterone synthetic enzymes in very preterm infants, impaired function in critically ill patients, and delayed recovery of the suppressed adrenal gland following contralateral adrenalectomy. Several medications may also impair synthesis, including angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, heparin, cyclosporine, tacrolimus, and ketoconazole.
Defective release
Defective release, or hyporeninemic hypoaldosteronism, reflects reduced renin or angiotensin II production. This form frequently arises in acquired conditions, eg, type 4 renal tubular acidosis, chronic kidney disease, diabetic nephropathy, autonomic neuropathy, sickle cell disease, and HIV infection. Medications, including beta blockers, nonsteroidal anti-inflammatory drugs, and dopamine, can further suppress aldosterone release.
Aldosterone resistance
Aldosterone resistance develops when aldosterone production remains intact but target tissues fail to respond appropriately. Genetic forms include autosomal dominant pseudohypoaldosteronism type 1, autosomal recessive pseudohypoaldosteronism type 1, and Gordon syndrome (pseudohypoaldosteronism type II). Acquired resistance may occur in pediatric urinary tract infections, obstructive uropathy (pseudohypoaldosteronism type III), or following the downregulation of mineralocorticoid receptors after solid organ transplantation. Medication-induced resistance often results from using mineralocorticoid receptor antagonists (eg, spironolactone or eplerenone).
Adrenal Gland Failure
Adrenal gland failure results from impaired development, destruction of adrenal tissue, or suppression of adrenal function. The condition may arise through genetic, acquired, or medication-induced mechanisms, each contributing to reduced aldosterone production and subsequent mineralocorticoid deficiency. Genetic causes include congenital adrenal hyperplasia, in which enzyme defects disrupt steroidogenesis, and adrenal hypoplasia, where underdevelopment of the adrenal cortex limits hormone production. Both conditions present early in life and require careful management to prevent life-threatening salt-wasting crises.
Acquired adrenal failure often develops through direct destruction of adrenal tissue. Autoimmune processes remain a leading cause, while bilateral adrenal hemorrhage, infectious adrenalitis caused by pathogens, eg, tuberculosis or cytomegalovirus, and infiltrative conditions, including malignancy or amyloidosis, can also compromise adrenal integrity. Adrenal suppression may occur in the absence of structural damage, most commonly as a consequence of medications. Pharmacologic agents (eg, etomidate, mirotane) or prolonged administration of exogenous glucocorticoids or mineralocorticoids suppress endogenous hormone production, leaving patients vulnerable to adrenal insufficiency once therapy is withdrawn.
Epidemiology
Acquired causes of hypoaldosteronism are observed to be more prevalent than congenital causes. Acquired hypoaldosteronism in adults is frequently attributed to hyporeninemic hypoaldosteronism, which often results from chronic diseases or the use of medications that interfere with RAAS. Congenital causes of hypoaldosteronism predominantly occur in specific ethnic groups, which may reflect founder effects and population-specific mutations.[10][11] The most common cause of congenital isolated hypoaldosteronism is aldosterone synthase deficiency (ASD), resulting from mutations in the CYP11B2 gene.[12] Autosomal dominant pseudohypoaldosteronism type I is another rare congenital cause of hypoaldosteronism, and has a reported prevalence of less than 1 in 80,000 live births.
Pathophysiology
Transient Pediatric Hypoaldosteronism
Preterm and very preterm neonates often develop a transient form of hypoaldosteronism that results from delayed maturation of adrenal synthetic pathways. These infants maintain intact renin stimulation, yet insufficient 11β-hydroxylase activity limits steroid conversion, and reduced mineralocorticoid receptor expression diminishes tissue responsiveness. This developmental immaturity contributes to impaired aldosterone-mediated regulation of sodium and potassium balance during the neonatal period. The condition generally resolves spontaneously as adrenal enzyme activity and receptor expression mature, with normalization occurring within the first few weeks to months of life.[13][14] Young individuals with urinary tract infections and obstructive uropathy may exhibit transient resistance to aldosterone, a condition known as pseudohypoaldosteronism III. The responsiveness of renal tubules to aldosterone activity typically improves following treatment of the underlying urologic condition.[15]
Persistent Pediatric Hypoaldosteronism
ASD is a hereditary condition linked to mutations in the CYP11B2 gene.[15] Over 40 mutations have been identified, most of which are either missense or nonsense. Autosomal dominant pseudohypoaldosteronism type 1 is caused by mutations in the NR3C2 gene, which encodes the mineralocorticoid receptor. At least 50 mutations have been associated with this condition, and the disease can be expressed with a single pathogenic allele. In contrast, autosomal recessive pseudohypoaldosteronism type 1 stems from loss-of-function mutations in the SCNN1A, SCNN1B, or SCNN1G genes, which encode the α, β, or γ subunits of epithelial sodium channels. This condition results in impaired assembly and expression of epithelial sodium channels.
Pseudohypoaldosteronism II is caused by mutations in the WNK1, WNK4, KLHL3, or CUL3 genes, resulting in gain-of-function mutations affecting the thiazide-sensitive sodium-chloride cotransporter (NCC). Ongoing research advances continue to uncover additional genes responsible for heritable hypoaldosteronism, eg, SEC61A1, which is involved in renin production.[16] These insights enhance our understanding of the molecular mechanisms underlying these conditions and may guide future therapeutic strategies.
Hyperreninemic Hypoaldosteronism
Hyperreninemic hypoaldosteronism occurs when impairment of aldosterone activity triggers a compensatory increase in plasma renin. Existing literature indicates that proinflammatory cytokines released during acute critical illness may suppress aldosterone production. A prospective study conducted in 2008 examined patients with liver cirrhosis who were hospitalized in the ICU. The findings demonstrated that disease severity and poor prognosis were positively correlated with elevated renin-to-aldosterone ratios and interleukin-6 levels.[17]
Unilateral adrenalectomy in patients with Conn syndrome can be associated with transient hyperreninemic hypoaldosteronism, attributable to the delayed recovery of the suppressed contralateral adrenal gland.[18][19] Angiotensin-converting enzyme inhibitors decrease angiotensin II production. Angiotensin receptor blockers (ARBs) block the stimulatory action of angiotensin II and prevent aldosterone secretion.
Conventional and low-molecular-weight heparin (LMWH) decrease aldosterone production and release; a similar effect is observed with cyclosporin and calcium channel blockers. LMWH diminishes the sensitivity of the zona glomerulosa to angiotensin II. Drugs like ketoconazole, which are also used in Cushing disease and other cancers, may affect all 3 axes of the adrenal synthetic pathways and diminish the action of angiotensin II.
Hyporeninemic Hypoaldosteronism
Hyporeninemic hypoaldosteronism, also referred to as type 4 RTA, develops when renin output decreases, leading to inadequate aldosterone production. The decline in renin disrupts sodium and potassium homeostasis, resulting in clinical and biochemical features consistent with mineralocorticoid deficiency.[20] Renin production depends heavily on sympathetic stimulation. Defective release frequently arises from autonomic neuropathies and becomes aggravated by renal disease, medications affecting the RAA axis, production of inactive renin, diminished beta-receptor sensitivity, and impaired prostaglandin synthesis.
Patients most often present in the sixth to eighth decades of life, commonly with diabetes, mild to moderate renal impairment, and concurrent use of drugs that interfere with the RAA system. Clinical manifestations frequently include asymptomatic hyperkalemia, hyponatremia, and hyperchloremic normal anion gap acidosis, with laboratory evaluation showing low serum aldosterone. In patients with HIV, viral adrenalitis, nephropathy, and the use of trimethoprim can also contribute to this disorder.
Several medications influence the renin and aldosterone pathways. Beta-blockers decrease renin release, while NSAIDs inhibit cyclooxygenase and blunt prostaglandin-mediated stimulation of renin. Spironolactone both suppresses aldosterone biosynthesis and competitively antagonizes the mineralocorticoid receptor. Amiloride and trimethoprim block epithelial sodium channels in renal tubules, increasing sodium excretion and promoting potassium retention. Triamterene acts as a potassium-sparing diuretic by targeting aldosterone-independent ion-exchange sites in the distal convoluted tubule. Dopamine exerts a tonic inhibitory effect on aldosterone production. High doses used in critical care settings, as well as therapy with dopamine agonists, can further suppress aldosterone synthesis and precipitate clinically significant hypoaldosteronism under certain circumstances.
History and Physical
Children with autosomal dominant pseudohypoaldosteronism typically exhibit a milder phenotype, presenting primarily with renal hyponatremia, hyperkalemia, and metabolic acidosis during infancy. Many affected individuals improve with age and often become asymptomatic after early childhood.[21] In contrast, autosomal recessive pseudohypoaldosteronism usually involves multiorgan manifestations, including recurrent respiratory tract infections, skin rashes, and inspissated secretions from the meibomian glands, in addition to the classic features of hypoaldosteronism.[22]
Prenatal detection of polyhydramnios may provide an early clue to autosomal recessive forms.[23] These children often require ongoing complex care and frequent hospitalizations, as symptoms persist throughout life. Patients with pseudohypoaldosteronism type III typically recover once underlying urinary tract infections or obstructive uropathy resolve.[15] In adult populations, careful review of medications remains essential, as rare cases of life-threatening hyperkalemia–induced arrhythmias have occurred in patients receiving conventional heparin.[24]
Clinicians should maintain a high index of suspicion in hospitalized or critically ill patients, as well as in those with diabetes, nephropathies, sickle cell disease, or HIV, to enable early recognition and prompt intervention. Adults undergoing adrenalectomy for unilateral aldosterone-producing adenoma (Conn syndrome) may experience prolonged suppression of the contralateral zona glomerulosa. Risk factors for delayed recovery include advanced age, long-standing hypertension, large adenoma size, preoperative and postoperative renal dysfunction, and preoperative nonsteroidal anti-inflammatory drug use.[25]
Evaluation
Diagnostic Laboratory Studies
Early and accurate diagnosis of infants presenting with a salt-wasting crisis depends on obtaining a blood sample while the patient is clinically or biochemically symptomatic and before initiating treatment. This specimen, referred to as the "critical sample," allows laboratory testing to avoid confounding effects from therapy. One sample aliquot is analyzed for serum electrolytes, aldosterone, 17-hydroxyprogesterone (17-OHP), and plasma renin activity or concentration. This initial panel guides further diagnostic evaluation.
A second aliquot is reserved for confirmatory testing, which may include serum cortisol and adrenocorticotropic hormone (ACTH) in suspected congenital adrenal hyperplasia, 18-hydroxycortisosterone (18-OHC)—low to normal in type 1 ASD and elevated in type 2 ASD—and urinary tetrahydro-aldosterone, decreased in type 1 and low-normal to normal in type 2 ASD. In infants with ASD but normal aldosterone concentrations, the plasma aldosterone concentration/plasma renin concentration ratio helps clarify the diagnosis of primary hypoaldosteronism.[26]
Some infants with pseudohypoaldosteronism may have falsely low aldosterone levels on assay testing, a phenomenon referred to as the “hook effect,” where very elevated aldosterone levels can paradoxically result in low levels on certain assay methods, potentially resulting in a misdiagnosis. The assay should be repeated with progressively diluted samples to avoid this and achieve high aldosterone levels accurately. Aldosterone measurement using serial sample dilutions can reveal markedly elevated levels, confirming the diagnosis of pseudohypoaldosteronism and preventing misdiagnosis as an ASD.[27]
Key biochemical tests that may be performed in the evaluation of hypoaldosteroneism include:
- Serum electrolytes
- Serum and urine aldosterone
- Serum cortisol and plasma ACTH
- Serum 17-OHP
- 18-OHC (low to normal in type 1 and high in type 2 aldosterone synthase deficiency)
- Urinary tetrahydroaldosterone, the metabolite of aldosterone (decreased in type 1 and low normal or normal in type 2 aldosterone synthase deficiency)
- Plasma renin activity or plasma renin concentration
- Plasma aldosterone concentration/plasma renin concentration ratio
- Plasma aldosterone concentration/potassium ratio (in chronic kidney disease, <10 ng/dL is suggestive of hypoaldosteronism and >10 ng/dL is suggestive of renal aldosterone resistance) [28]
- Serum creatinine and estimated glomerular filtration rate
- Molecular genetic studies, including gene sequencing (see Image. Genetics of Hypoaldosteronism)
Treatment / Management
Management Approach in Pediatric Populations
For pediatric individuals presenting with a salt-wasting crisis due to ASD, autosomal dominant pseudohypoaldosteronism, or autosomal recessive pseudohypoaldosteronism, the initial management entails aggressive rehydration with isotonic fluids. Before a definitive diagnosis of hypoaldosteronism is made, after drawing a critical sample of blood, patients should be empirically treated for presumed congenital adrenal hyperplasia with intravenous hydrocortisone in stress doses. This also has adequate mineralocorticoid activity and will tide over the crises. If 17-OHP levels are within normal range and a diagnosis of ASD has been made, fludrocortisone can be substituted orally for hydrocortisone after the infant stabilizes. Initial dosing of fludrocortisone for patients with ASD starts at 150 µg/m2 body surface area. The younger the child, the greater the doses of fludrocortisone because of drug insensitivity in infants and young children.
Fludrocortisone is ineffective as maintenance therapy in autosomal dominant pseudohypoaldosteronism and autosomal recessive pseudohypoaldosteronism due to end-organ resistance. In these cases, the management approach focuses on supportive care, with aggressive correction of hyponatremia and hyperkalemia using high-dose sodium chloride supplementation and cation exchange resins. In severe or refractory cases, peritoneal dialysis, thiazide diuretics, and indomethacin may be useful.[29][30](B3)
Etiology-Guided Management
In Conn syndrome, discontinuing spironolactone and correction of any volume expansion for at least 3 days preoperatively is recommended. Following adrenalectomy, hypoaldosteronism is managed with fludrocortisone. Some patients may require extended support until their adrenal function fully recovers from suppression.[31][32]
Renal transplant individuals treated with calcineurin inhibitors present with hyperkalemia secondary to hypoaldosteronism and respond favorably to a low dose of fludrocortisone.[33] In type 4 renal tubular acidosis, fludrocortisone is useful in reducing hyperkalemia, but caution is required as sodium retention, fluid overload, and precipitation of congestive heart failure are potential adverse effects. Adding a loop diuretic, or even better, thiazides that block the sodium-chloride cotransporter in the distal renal tubules, may be necessary. Precipitating medications, eg, nonsteroidal anti-inflammatory drugs and potassium-sparing diuretics, should be carefully reviewed and discontinued.(B2)
Differential Diagnosis
Hypoaldosteronism can be differentiated from global adrenal failure by the presence of genital ambiguity in female infants and a high 17-OHP in both sexes in congenital adrenal hyperplasia (CAH). Addison disease in older children and adults will have decreased serum cortisol and increased ACTH, present dramatically, and have physical findings like hyperpigmentation. Infants with CAH typically present with salt-wasting and hyperpigmentation, while cortisol deficiency usually develops later. A cosyntropin stimulation test may be necessary to confirm cortisol deficiency. Deficiency of cholesterol side-chain cleaving enzymes produces deficiencies in all 3 adrenal axes. Thus, salt-wasting, low cortisol, and ambiguous genitalia in male infants are seen.
Decreased aldosterone and increased plasma renin concentration are features of aldosterone synthase deficiency. In contrast, pseudohypoaldosteronism is characterized by a very high plasma renin concentration and aldosterone, although both conditions exhibit low sodium, high potassium, and acidosis. A definitive diagnosis of biosynthetic defects causing hypoaldosteronism is typically made through gene sequencing.[12]
A preoperative aldosterone contralateral suppression index (see Image. Adrenal Venous Sampling) during adrenal venous sampling is the ratio of aldosterone to cortisol in the ipsilateral adrenal vein divided by a similar ratio in the external iliac vein. Patients with a contralateral suppression index of less than 0.47 are at risk for prolonged adrenal suppression and require close monitoring of serum potassium after unilateral adrenalectomy for Conn syndrome.
In critically ill individuals, hypoaldosteronism often lacks clinical significance because elevated cortisol levels activate the mineralocorticoid receptor, compensating for reduced aldosterone activity. Certain conditions, however, present with low aldosterone levels secondary to sodium retention, hypertension, hypokalemia, suppressed plasma renin concentration, and extracellular volume expansion. Based on these characteristic features, these states must be distinguished from salt-wasting causes of hypoaldosteronism. Examples include Liddle syndrome and the syndrome of mineralocorticoid excess, caused by the inhibition of 11β-hydroxysteroid dehydrogenase type 2. Gordon syndrome represents another distinct entity, in which patients exhibit hypertension accompanied by hyperkalemia, hyperchloremic acidosis, and suppressed renin levels, all resulting from chronic volume expansion. Accurate differentiation of these conditions is essential to guide appropriate management and avoid unnecessary interventions.
Prognosis
The prognosis of the congenital forms depends on the subtype. Patients with aldosterone synthase deficiency and autosomal dominant pseudohypoaldosteronism can be weaned off medications and even go into remission if identified early and promptly treated. Older children who are inadequately treated have retarded growth or failure to thrive, but adequately treated children can grow normally and have a good catch-up growth.
Autosomal recessive pseudohypoaldosteronism patients, as a rule, do not improve and require close monitoring. Pseudohypoaldosteronism III patients will be cured once their primary renal infection or obstruction is resolved. Diligent management of medications causing hypoaldosteronism will reduce morbidity considerably. Most patients with hypoaldosteronism postadrenalectomy for Conn syndrome resolve early, while some at high risk may need longer than 6-month surveillance.
Complications
In patients treated for hypoaldosteronism, overdosage with fludrocortisone can cause fluid retention, low potassium, significantly suppressed plasma renin concentration, and, ultimately, can even precipitate congestive heart failure. Less severe but clinically significant adverse effects include constipation due to increased colonic mucosal sodium and water reabsorption, weight gain due to fluid retention, and muscle weakness due to hypokalemia. Dosing should be titrated to plasma renin concentration levels in the upper limits of the normal range. Undertreated children will have poor growth. When the diagnosis is delayed in infants with severe disease, mortality is increased.
Consultations
The following clinicians may be involved in the management of hypoaldosteronism:
- Endocrinology (pediatric and adult)
- Nephrology (pediatric and adult)
- Emergency medicine (pediatric and adult)
- Laboratory medicine
- Molecular genetics
- Clinical psychology
- Nursing
Deterrence and Patient Education
Deterrence in hypoaldosteronism focuses on preventing complications related to electrolyte imbalances, fluid dysregulation, and cardiovascular risk. Clinicians should proactively monitor at-risk populations, including preterm infants, patients with chronic kidney disease, diabetes, adrenal insufficiency, or those receiving medications that impair aldosterone synthesis or action. Early recognition of biochemical abnormalities, eg, hyponatremia, hyperkalemia, and metabolic acidosis, allows timely interventions, reducing the likelihood of life-threatening events, eg, arrhythmias, severe dehydration, or salt-wasting crises. Medication review plays a critical role, as agents like nonsteroidal anti-inflammatory drugs, beta-blockers, and mineralocorticoid receptor antagonists can exacerbate hypoaldosteronism and contribute to preventable complications.
Patient education empowers families and patients to manage and prevent diseases actively. Caregivers of neonates or children with congenital or genetic forms, such as aldosterone synthase deficiency or pseudohypoaldosteronism, should understand the importance of fluid and electrolyte replacement, adherence to medications like fludrocortisone, and monitoring for early signs of imbalance. Adults with this condition should be informed about potential medication interactions, the risks of hyperkalemia, and strategies to maintain appropriate sodium and potassium balance. Education promotes adherence, encourages prompt reporting of concerning symptoms, and supports long-term outcomes by reducing hospitalizations and enhancing safety.
Pearls and Other Issues
Hypoaldosteronism in children is rare, but proper and timely recognition can be life-saving. Many advances have elucidated the mechanisms in the etiology, pathophysiology, genetics, evaluation, and management of hypoaldosteronism, and a better understanding of these concepts will help reduce morbidity and mortality. Hypoaldosteronism can occur isolated or as part of generalized adrenal failure.
In infancy, the presentation is often dramatic with a salt-wasting crisis; older children present with failure to thrive or poor growth. A high index of suspicion is required in children with hyperkalemia, particularly when a negative CAH screen is present. Hyperkalemia in the absence of renal insufficiency may indicate the presence of hypoaldosteronism.
Aldosterone synthase deficiency is typically treated with sodium chloride and fludrocortisone and has a favorable prognosis. Autosomal dominant pseudohypoaldosteronism and autosomal recessive pseudohypoaldosteronism resist fludrocortisone; the former has a good prognosis, while the latter has a poor outcome due to generalized disease. Identification of high-risk individuals will aid in better management of postadrenalectomy complications in Conn syndrome. The knowledge that various medications, including over-the-counter and alternative medicines, may cause hypoaldosteronism will improve patient outcomes. High-risk patients with diabetes and mild renal dysfunction are prone to type 4 renal tubular acidosis. Care is required in diligently avoiding medications like potassium-sparing diuretics, nonsteroidal anti-inflammatory drugs, and trimethoprim.
Enhancing Healthcare Team Outcomes
Hypoaldosteronism encompasses a spectrum of disorders characterized by insufficient aldosterone synthesis, impaired release, or end-organ resistance, resulting in disrupted sodium and potassium balance, fluid dysregulation, and metabolic disturbances. In pediatric populations, severe manifestations include salt-wasting crises that require urgent recognition and management, whereas adults may present with hyperkalemia, hypertension, or complications from renal or adrenal pathology. Etiologies range from congenital enzyme deficiencies and genetic syndromes such as pseudohypoaldosteronism to medication-induced suppression and acquired adrenal failure. Accurate diagnosis relies on timely biochemical testing, critical sampling, and, when indicated, genetic analysis. Management focuses on targeted interventions, such as fludrocortisone therapy, electrolyte replacement, and correction of reversible causes. Early recognition and individualized care reduce morbidity and mortality and support long-term growth and development in children.
Effective management of hypoaldosteronism requires coordinated, interprofessional care. Pediatric emergency clinicians initiate stabilization during salt-wasting crises, after which general pediatricians assume ongoing care, incorporating periodic guidance from pediatric endocrinologists. Clinical geneticists aid in confirming specific diagnoses, while pediatric nephrologists intervene when hyperkalemia necessitates dialysis. Nurses and clinical psychologists are essential in monitoring, educating, and supporting families through complex care processes. In adults, Conn syndrome management demands collaboration among endocrinologists, endocrine surgeons, interventional radiologists, laboratory personnel, and nurses skilled in postoperative care. Patients with type 4 renal tubular acidosis benefit from primary care oversight coordinated with nephrology specialists. Across all populations, effective communication, shared decision-making, and coordinated care enhance patient safety, optimize outcomes, and strengthen overall team performance.
Media
(Click Image to Enlarge)

Adrenal Venous Sampling. Adrenal venous sampling is the ratio of aldosterone to cortisol in the ipsilateral adrenal vein divided by a similar ratio in the external iliac vein.
Contributed by V Rajkumar, MD
(Click Image to Enlarge)
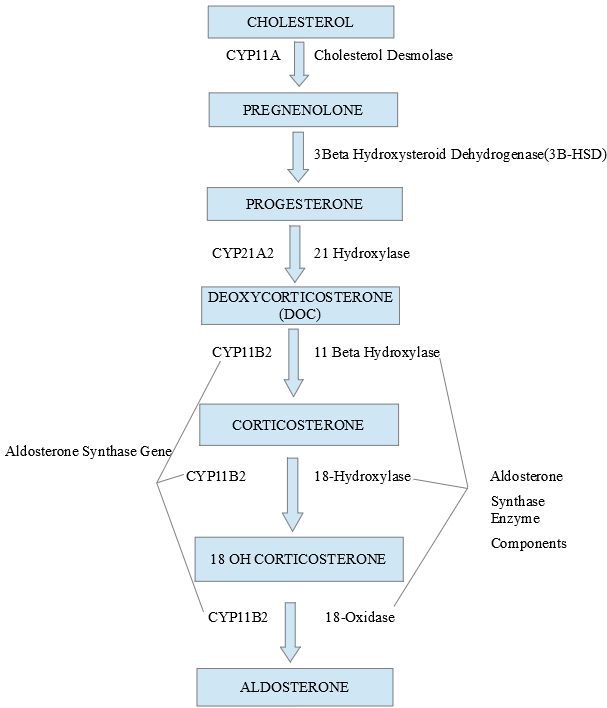
Aldosterone Synthase Enzyme. Image showing components of the aldosterone synthase enzyme and the multiple steps it catalyzes to produce aldosterone.
Contributed by V Rajkumar, MD
(Click Image to Enlarge)
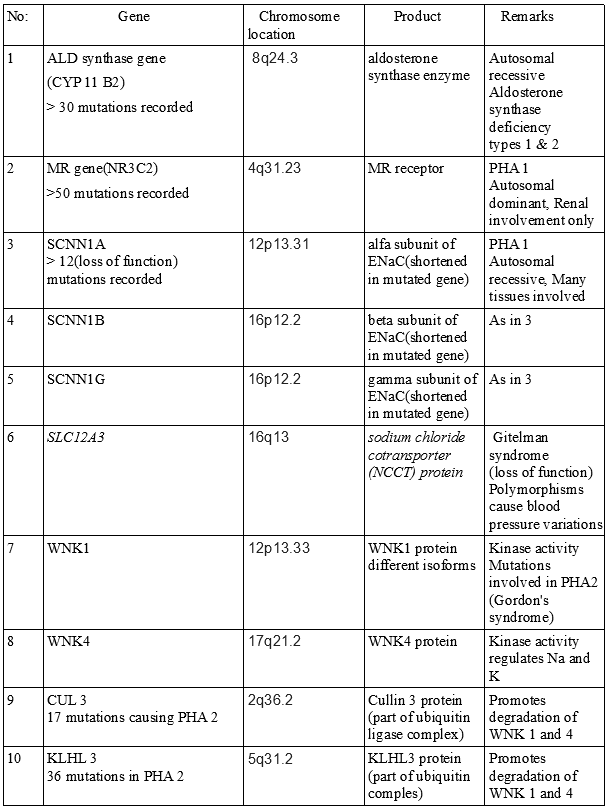
Genetics of Hypoaldosteronism. Molecular genetic studies can help identify hypoaldosteronism and pseudohypoaldosteronism.
Contributed by V Rajkumar, MD
References
Calissendorff J, Falhammar H. Renal pseudohypoaldosteronism type 1-an adult case series including a novel gene variant. Endocrine. 2025 Mar:87(3):1285-1290. doi: 10.1007/s12020-024-04120-8. Epub 2024 Nov 29 [PubMed PMID: 39614070]
Level 2 (mid-level) evidenceÅkerström T, Klimàcek B, Annebäck M, Norlén O, Stålberg P. Intratumoural aldosterone and CYP11B2 expression levels among different genotypes of aldosterone producing tumours. Endocrine connections. 2025 Jun 1:14(6):. pii: e250070. doi: 10.1530/EC-25-0070. Epub 2025 Jun 26 [PubMed PMID: 40531063]
Wang L, Zhang Z, Liu D, Yuan K, Zhu G, Qi X. Association of -344C/T polymorphism in the aldosterone synthase (CYP11B2) gene with cardiac and cerebrovascular events in Chinese patients with hypertension. The Journal of international medical research. 2020 Sep:48(9):300060520949409. doi: 10.1177/0300060520949409. Epub [PubMed PMID: 32938270]
Level 2 (mid-level) evidenceVamsi UM, Swapna N, Padma G, Vishnupriya S, Padma T. Haplotype association and synergistic effect of human aldosterone synthase (CYP11B2) gene polymorphisms causing susceptibility to essential hypertension in Indian patients. Clinical and experimental hypertension (New York, N.Y. : 1993). 2016:38(8):659-665. doi: 10.1080/10641963.2016.1200595. Epub 2016 Dec 9 [PubMed PMID: 27935319]
Barrett PQ, Guagliardo NA, Bayliss DA. Ion Channel Function and Electrical Excitability in the Zona Glomerulosa: A Network Perspective on Aldosterone Regulation. Annual review of physiology. 2021 Feb 10:83():451-475. doi: 10.1146/annurev-physiol-030220-113038. Epub 2020 Nov 11 [PubMed PMID: 33176563]
Level 3 (low-level) evidenceNanba K, Chen A, Nishimoto K, Rainey WE. Role of Ca(2+)/calmodulin-dependent protein kinase kinase in adrenal aldosterone production. Endocrinology. 2015 May:156(5):1750-6. doi: 10.1210/en.2014-1782. Epub 2015 Feb 13 [PubMed PMID: 25679868]
El Ghorayeb N, Bourdeau I, Lacroix A. Role of ACTH and Other Hormones in the Regulation of Aldosterone Production in Primary Aldosteronism. Frontiers in endocrinology. 2016:7():72. doi: 10.3389/fendo.2016.00072. Epub 2016 Jun 27 [PubMed PMID: 27445975]
Abedini A, Sánchez-Navaro A, Wu J, Klötzer KA, Ma Z, Poudel B, Doke T, Balzer MS, Frederick J, Cernecka H, Liu H, Liang X, Vitale S, Kolkhof P, Susztak K. Single-cell transcriptomics and chromatin accessibility profiling elucidate the kidney-protective mechanism of mineralocorticoid receptor antagonists. The Journal of clinical investigation. 2024 Jan 2:134(1):. doi: 10.1172/JCI157165. Epub 2024 Jan 2 [PubMed PMID: 37906287]
Tomkins M, McDonnell T, Cussen L, Sagmeister MS, Oestlund I, Shaheen F, Harper L, Hardy RS, Taylor AE, Gilligan LC, Arlt W, McIlroy M, de Freitas D, Conlon P, Magee C, Denton M, O'Seaghdha C, Snoep JL, Storbeck KH, Sherlock M, O'Reilly MW. Impaired 11β-Hydroxysteroid Dehydrogenase Type 2 Activity in Kidney Disease Disrupts 11-Oxygenated Androgen Biosynthesis. The Journal of clinical endocrinology and metabolism. 2025 May 19:110(6):1701-1715. doi: 10.1210/clinem/dgae714. Epub [PubMed PMID: 39382395]
Hanukoglu A, Vargas-Poussou R, Landau Z, Yosovich K, Hureaux M, Zennaro MC. Renin-aldosterone system evaluation over four decades in an extended family with autosomal dominant pseudohypoaldosteronism due to a deletion in the NR3C2 gene. The Journal of steroid biochemistry and molecular biology. 2020 Nov:204():105755. doi: 10.1016/j.jsbmb.2020.105755. Epub 2020 Oct 2 [PubMed PMID: 33017655]
Hubert EL, Teissier R, Fernandes-Rosa FL, Fay M, Rafestin-Oblin ME, Jeunemaitre X, Metz C, Escoubet B, Zennaro MC. Mineralocorticoid receptor mutations and a severe recessive pseudohypoaldosteronism type 1. Journal of the American Society of Nephrology : JASN. 2011 Nov:22(11):1997-2003. doi: 10.1681/ASN.2011030245. Epub 2011 Sep 8 [PubMed PMID: 21903996]
Merakou C, Fylaktou I, Sertedaki A, Dracopoulou M, Voutetakis A, Efthymiadou A, Christoforidis A, Dacou-Voutetakis C, Chrysis D, Kanaka-Gantenbein C. Molecular Analysis of the CYP11B2 Gene in 62 Patients with Hypoaldosteronism Due to Aldosterone Synthase Deficiency. The Journal of clinical endocrinology and metabolism. 2021 Jan 1:106(1):e182-e191. doi: 10.1210/clinem/dgaa765. Epub [PubMed PMID: 33098647]
Viengchareun S, Pussard E, Castanet M, Sachs LM, Vu TA, Boileau P, Lombès M, Martinerie L. The invention of aldosterone, how the past resurfaces in pediatric endocrinology. Molecular and cellular endocrinology. 2021 Sep 15:535():111375. doi: 10.1016/j.mce.2021.111375. Epub 2021 Jun 29 [PubMed PMID: 34197901]
Martinerie L, Viengchareun S, Delezoide AL, Jaubert F, Sinico M, Prevot S, Boileau P, Meduri G, Lombès M. Low renal mineralocorticoid receptor expression at birth contributes to partial aldosterone resistance in neonates. Endocrinology. 2009 Sep:150(9):4414-24. doi: 10.1210/en.2008-1498. Epub 2009 May 28 [PubMed PMID: 19477942]
Betti C, Lavagno C, Bianchetti MG, Kottanattu L, Lava SAG, Schera F, Lacalamita MC, Milani GP. Transient secondary pseudo-hypoaldosteronism in infants with urinary tract infections: systematic literature review. European journal of pediatrics. 2024 Oct:183(10):4205-4214. doi: 10.1007/s00431-024-05676-3. Epub 2024 Jul 10 [PubMed PMID: 38985174]
Level 1 (high-level) evidenceKarpman D, Lindström ML, Möller M, Ivarsson S, Kristoffersson AC, Bekassy Z, Fogo AB, Elfving M. Hypoaldosteronism due to a novel SEC61A1 variant successfully treated with fludrocortisone. Clinical kidney journal. 2024 Aug:17(8):sfae213. doi: 10.1093/ckj/sfae213. Epub 2024 Jul 5 [PubMed PMID: 39135939]
du Cheyron D, Bouchet B, Cauquelin B, Guillotin D, Ramakers M, Daubin C, Ballet JJ, Charbonneau P. Hyperreninemic hypoaldosteronism syndrome, plasma concentrations of interleukin-6 and outcome in critically ill patients with liver cirrhosis. Intensive care medicine. 2008 Jan:34(1):116-24 [PubMed PMID: 17906854]
Queiroz NL, Stumpf MAM, Souza VCM, Maciel AAW, Fagundes GFC, Okubo J, Srougi V, Tanno FY, Chambo JL, Pereira MAA, Pio-Abreu A, Bortolotto LA, Latronico AC, Barisson Villares Fragoso MC, Drager LF, Mendonça BB, Almeida MQ. Renal Function Evolution and Hypoaldosteronism Risk After Unilateral Adrenalectomy for Primary Aldosteronism. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2024 May:56(5):350-357. doi: 10.1055/a-2221-3302. Epub 2023 Dec 1 [PubMed PMID: 38040032]
Starker LF, Christakis I, Julien JS, Schwarz K, Graham P, Grubbs EG, Lee JE, Perrier ND. Considering Postoperative Functional Hypoaldosteronism after Unilateral Adrenalectomy. The American surgeon. 2017 Jun 1:83(6):598-604 [PubMed PMID: 28637561]
Palmer BF, Kelepouris E, Clegg DJ. Renal Tubular Acidosis and Management Strategies: A Narrative Review. Advances in therapy. 2021 Feb:38(2):949-968. doi: 10.1007/s12325-020-01587-5. Epub 2020 Dec 26 [PubMed PMID: 33367987]
Level 3 (low-level) evidenceGeller DS, Zhang J, Zennaro MC, Vallo-Boado A, Rodriguez-Soriano J, Furu L, Haws R, Metzger D, Botelho B, Karaviti L, Haqq AM, Corey H, Janssens S, Corvol P, Lifton RP. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. Journal of the American Society of Nephrology : JASN. 2006 May:17(5):1429-36 [PubMed PMID: 16611713]
Nur N, Lang C, Hodax JK, Quintos JB. Systemic Pseudohypoaldosteronism Type I: A Case Report and Review of the Literature. Case reports in pediatrics. 2017:2017():7939854. doi: 10.1155/2017/7939854. Epub 2017 Apr 18 [PubMed PMID: 28484659]
Level 3 (low-level) evidenceHuneif MA, Alhazmy ZH, Shoomi AM, Alghofely MA, Heena H, Mushiba AM, AlSaheel A. A Novel SCNN1A Variation in a Patient with Autosomal-recessive Pseudohypoaldosteronism Type 1. Journal of clinical research in pediatric endocrinology. 2022 Jun 7:14(2):244-250. doi: 10.4274/jcrpe.galenos.2021.2020.0175. Epub 2021 Apr 8 [PubMed PMID: 33829730]
Kovacs J, Talib S, Khashan A, Garsondiya B, Carson MP. A 77-Year-Old Man with Heparin-Induced Aldosterone Suppression Causing Hyperkalemia. The American journal of case reports. 2022 Jul 21:23():e937017. doi: 10.12659/AJCR.937017. Epub 2022 Jul 21 [PubMed PMID: 35859349]
Level 3 (low-level) evidenceTahir A, McLaughlin K, Kline G. Severe hyperkalemia following adrenalectomy for aldosteronoma: prediction, pathogenesis and approach to clinical management- a case series. BMC endocrine disorders. 2016 Jul 27:16(1):43. doi: 10.1186/s12902-016-0121-y. Epub 2016 Jul 27 [PubMed PMID: 27460219]
Level 2 (mid-level) evidenceRuecker B, Lang-Muritano M, Spanaus K, Welzel M, l'Allemand D, Phan-Hug F, Katschnig C, Konrad D, Holterhus PM, Schoenle EJ. The Aldosterone/Renin Ratio as a Diagnostic Tool for the Diagnosis of Primary Hypoaldosteronism in Newborns and Infants. Hormone research in paediatrics. 2015:84(1):43-8. doi: 10.1159/000381852. Epub 2015 May 9 [PubMed PMID: 25968592]
Ghazal K, Brabant S, Prie D, Piketty ML. Hormone Immunoassay Interference: A 2021 Update. Annals of laboratory medicine. 2022 Jan 1:42(1):3-23. doi: 10.3343/alm.2022.42.1.3. Epub [PubMed PMID: 34374345]
Kim M, Lee HY, Kim H. Impaired aldosterone response to potassium and hyperkalemia in patients receiving a renin-angiotensin-aldosterone system inhibitor. The Korean journal of internal medicine. 2025 May:40(3):468-481. doi: 10.3904/kjim.2024.160. Epub 2025 Apr 30 [PubMed PMID: 40360223]
Gurpinar Tosun B, Kendir Demirkol Y, Seven Menevse T, Kaygusuz SB, Ozbek MN, Altincik SA, Mammadova J, Cayir A, Doger E, Bayramoglu E, Nalbantoglu O, Yesiltepe Mutlu G, Aghayev A, Turan S, Bereket A, Guran T. Catch-up Growth and Discontinuation of Fludrocortisone Treatment in Aldosterone Synthase Deficiency. The Journal of clinical endocrinology and metabolism. 2022 Jan 1:107(1):e106-e117. doi: 10.1210/clinem/dgab619. Epub [PubMed PMID: 34415991]
Alkhatib EH, Bartlett D, Kanakatti Shankar R, Regier D, Merchant N. Case report: Early molecular confirmation and sodium polystyrene sulfonate management of systemic pseudohypoaldosteronism type I. Frontiers in endocrinology. 2023:14():1297335. doi: 10.3389/fendo.2023.1297335. Epub 2024 Jan 15 [PubMed PMID: 38288475]
Level 3 (low-level) evidencePreda C, Teodoriu LC, Placinta S, Grigorovici A, Bilha S, Ungureanu CM. Persistent severe hyperkalemia following surgical treatment of aldosterone-producing adenoma. Journal of research in medical sciences : the official journal of Isfahan University of Medical Sciences. 2020:25():17. doi: 10.4103/jrms.JRMS_603_19. Epub 2020 Feb 20 [PubMed PMID: 32174989]
Mermejo LM, Elias PCL, Molina CAF, Tucci S, Muglia VF, Elias J, Antonini SR, de Castro M, Moreira AC. Early Renin Recovery After Adrenalectomy in Aldosterone-Producing Adenomas: A Prospective Study. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2022 Apr:54(4):224-231. doi: 10.1055/a-1778-4002. Epub 2022 Apr 12 [PubMed PMID: 35413743]
Gama RM, Makanjuola D, Wahba M, Quan V, Phanish M. Fludrocortisone Is an Effective Treatment for Hyperkalaemic Metabolic Acidosis in Kidney Transplant Recipients on Tacrolimus: A Case Series. Nephron. 2022:146(2):190-196. doi: 10.1159/000519670. Epub 2021 Nov 16 [PubMed PMID: 34784594]
Level 2 (mid-level) evidence