Introduction
Hyperammonemia is a life-threatening metabolic disturbance characterized by elevated plasma ammonia, a neurotoxic nitrogenous compound. Ammonia is primarily generated from intestinal bacterial metabolism and amino acid catabolism, enters the portal circulation, and is normally detoxified in the liver via the urea cycle.[1] Impairment of this pathway due to urea cycle disorders, hepatocellular dysfunction, or portosystemic shunting results in systemic accumulation and neuronal injury.[2] Normal plasma ammonia levels vary with age, being highest in neonates. Clinically, hyperammonemia is defined as more than 100 μmol/L in neonates and more than 50 μmol/L in older children and adults.[2] Concentrations exceeding these thresholds rapidly disrupt cerebral energy metabolism and osmoregulation, leading to encephalopathy, cerebral edema, seizures, coma, and potentially death.
In adults, approximately 90% of cases are related to advanced liver disease, most commonly cirrhosis or acute liver failure.[1][2] Nonhepatic causes include urea cycle disorders and other inborn errors of metabolism, infections with urease-producing bacteria, medication-induced hyperammonemia (eg, valproate, select chemotherapeutics), and catabolic stressors such as gastrointestinal bleeding, starvation, surgery, or trauma.[1][3] Because neurologic injury can progress within hours, early recognition and immediate intervention are critical. Initial management focuses on rapid ammonia reduction and suppression of catabolism while the underlying precipitant is identified and addressed.[1] Optimal care requires coordinated involvement of hepatology, medical genetics and metabolism, neurology, nephrology, and nutrition.[1]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Hyperammonemia results from impaired hepatic detoxification or excess ammonia production. Causes are congenital (inborn errors of metabolism) or acquired (hepatic and non-hepatic). Early mechanism identification guides emergent therapy and long-term management (see Image. Hyperammonemia, Laboratory Diagnosis Table).
Pediatric Causes
Congenital causes (inborn errors of metabolism)
- Urea cycle disorders: Pathogenic variants in NAGS, CPS1, OTC, ASS1, ASL, or ARG1 reduce urea formation. Inheritance is autosomal recessive, except for ornithine transcarbamylase (OTC), which is X-linked.[4][5][6]
- Proximal defects (NAGS/CPS1/OTC): Classically present at 24 to 48 hours with lethargy, vomiting, seizures, and encephalopathy [7][8]
- Arginase (ARG1) deficiency: Typically milder hyperammonemia; signs are spasticity from hyperargininemia [9][10]
- Transient hyperammonemia of the newborn (THAN): Predominantly in preterm infants due to immature urea-cycle function and hepatic hypoxia/microthrombosis; onset at 24 to 48 hours
- Mimics urea cycle disorders but often resolves with supportive care (dialysis if severe) [11]
Nonurea cycle genetic causes
- Organic acidemias (propionic, methylmalonic): CPS1 inhibition; often with metabolic acidosis/ketosis
- Fatty-acid oxidation defects (medium-chain acyl-coenzyme A [CoA] dehydrogenase; late-onset multiple acyl-CoA dehydrogenase deficiency [MADD]): Hyperammonemic encephalopathy possible; some MADD cases are riboflavin-responsive [12]
- Transporter/mitochondrial disorders: HHH (SLC25A15); lysinuric protein intolerance (SLC7A7); citrin deficiency (adult-onset citrullinemia type II); carbonic anhydrase VA deficiency (may respond to carglumic acid); hyperinsulinism–hyperammonemia (GLUD1 ± SLC25A36) [13][14]
- Others (rare): Adult-onset 3-methylcrotonyl-CoA carboxylase deficiency; selected mitochondrial DNA depletion syndromes [7]
Perinatal causes
These causes include severe perinatal asphyxia and neonatal herpes simplex virus with hepatic failure.[15]
Reye syndrome
Reye syndrome is a rare but serious condition that typically affects children and adolescents recovering from a viral infection, such as influenza or varicella (chickenpox). This condition is often associated with aspirin use during the viral illness, though the exact mechanism isn’t fully understood.
Acquired Causes
Hepatic causes (hepatic encephalopathy)
- Type A: Acute liver failure (viral, ischemic, toxins)
- Type B: Portosystemic shunting without intrinsic liver disease (congenital or post-surgical)
- Type C: Cirrhosis with portal hypertension (eg, hepatitis B and C viruses, Wilson disease, alpha-1 antitrypsin deficiency, cystic fibrosis)
- Common precipitants: Renal failure, gastrointestinal bleeding, infection, and dehydration [1][16]
Nonhepatic causes
- Hematologic malignancy/therapy: Acute lymphocytic leukemia; myeloma; asparaginase commonly causes transient hyperammonemia; severe cases require intensive care unit care and ammonia-lowering therapy.
- Infections, urease producers: Proteus, Klebsiella, Morganella, Providencia, Staphylococcus saprophyticus (often with urinary obstruction/retention)
- Infections, nonurease mechanisms: Actinotignum schaalii urinary tract infection, severe pneumonia with ammonia-producing airway flora, pleural empyema, and pylephlebitis
- Unmasked urea cycle disorders: Partial defects can decompensate with illness, surgery, total parenteral nutrition, pregnancy/postpartum, or after bariatric surgery
- Drugs/toxins: Valproate (risk increased with topiramate, high dose, liver disease, carnitine deficiency), valproate–topiramate combination, 5-fluorouracil ± platinum; others include carbamazepine, ribavirin, sulfadiazine, salicylates, amiodarone, endoxifen, deferasirox (via Fanconi syndrome), L-asparaginase [3][21]
- Procedure-related: Operative hysteroscopy with glycine absorption causing hypo-osmolar hyponatremia and hyperammonemia; correct fluid/solute imbalance
- Systemic states: Renal failure, severe malnutrition, gastrointestinal bleeding, high tumor burden, severe dehydration (transient elevations normalize with rehydration) [1][16]
- Transplant recipients: Lung and hematopoietic stem cell transplant recipients are vulnerable to fulminant, often infection-related, hyperammonemia with high morbidity/mortality [22]
- Adults without liver disease: Consider urease-producing infections, valproate (± topiramate), hematologic malignancy/therapy, and vascular anomalies.[19][23][24][25][26]
Epidemiology
Hyperammonemia patterns vary by age and etiology. In adults, most cases stem from advanced liver disease or portosystemic shunting; in neonates/children, inborn errors of metabolism dominate non-hepatic causes.[11][26]
- Urea cycle disorders: Historical incidence ~1:35,000 live births; contemporary estimates up to ~1:8200 in the United States. Ornithine transcarbamylase, a key enzyme in the urea cycle, responsible for detoxifying ammonia in the liver, is the most common single defect.[5][6][27]
- Screening: ASS1/ASL defects and several organic acidemias are on standard newborn screens (NBS); ornithine transcarbamylase and N-acetylglutamate synthase are typically not part of NBS; therefore, a normal NBS does not exclude a urea cycle disorder in symptomatic infants.[28]
- Age/presentation: Early-onset disease presents at 24 to 72 hours with severe hyperammonemia.[29] Partial/late-onset forms may first appear in childhood or adulthood, often triggered by infection, surgery, pregnancy/postpartum, total parenteral nutrition, or post-bariatric states.[30][31]
Pathophysiology
Hyperammonemia occurs when ammonia production exceeds hepatic detoxification. Portal venous ammonia is normally converted to urea in the liver. Extrahepatic tissues (skeletal muscle, brain, kidney) buffer nitrogen by forming glutamine and alanine, but this sink is quickly overwhelmed during acute or sustained elevations.[32][33][34] See Image. Urea Cycle.
Sources of Ammonia (Amino Acid Catabolism)
- Gut/colon: Bacterial urease and protein metabolism [35]
- Small intestine: Enterocyte glutaminase (augmented by bacterial overgrowth)
- Skeletal muscle: Amino-acid transamination and the purine-nucleotide cycle [34]
- The amino acids that directly produce ammonia are glutamine, glutamate, and asparagine
- Aspartate, glutamate, glutamine, and the urea cycle amino acids are also especially important [36]
Central Nervous System Mechanisms
Astrocyte swelling leading to cerebral edema represents the core pathway of hyperammonemic neurotoxicity. Ammonia readily crosses the blood–brain barrier, where astrocytic glutamine synthetase converts ammonia and glutamate into glutamine. Intracellular accumulation of glutamine acts as an osmolyte, driving cytotoxic edema. This cascade results in mitochondrial dysfunction, enhanced oxidative and nitrosative stress, disrupted potassium and water homeostasis, and activation of programmed cell death pathways.[32]
Neurotransmitter dysregulation
- Glutamatergic excitotoxicity arises when excess extracellular glutamate overactivates N-methyl-D-aspartate receptors, increasing sodium–potassium adenosine triphosphatase demand, depleting cellular adenosine triphosphate, and ultimately precipitating seizures and neuronal injury.[37][38]
- Chronic adaptations: Persistent extrasynaptic glutamate impairs receptor signaling and the glutamate–NO–cyclic guanosine monophosphate pathway, driving cognitive deficits in hepatic encephalopathy.[39]
- Gamma-aminobutyric acid (GABA) GABAergic tone: Endogenous benzodiazepine-like ligands/neurosteroids enhance inhibition and depress synaptic activity.[40][41]
- Serotonergic changes: Altered tryptophan transport can raise central serotonin and contribute to anorexia in chronic hyperammonemia.[42]
Structural Injury and Biomarkers
Chronic or recurrent hyperammonemia is linked to characteristic white-matter injury, often involving corticospinal tracts. Advanced neuroimaging (eg, diffusion tensor imaging) can detect microstructural changes and is being explored as a biomarker of disease burden and recovery in uric cycle disorder (UCDs).[43][35]
Exposure Dynamics
In UCDs, neurotoxicity reflects astrocytic glutamine accumulation, oxidative stress, and impaired neurotransmission. Rapid rises in ammonia correlate more strongly with acute brain injury than cumulative exposure alone.[35]
Hepatic and Systemic Effects
Ammonia is both neurotoxic and hepatotoxic; it promotes oxidative stress, mitochondrial permeability transition, apoptosis/necroptosis, and fibrogenesis, worsening hepatic dysfunction and perpetuating hyperammonemia.[44][45] Within the gut–liver–brain axis, ammonia drives liver injury and hepatic encephalopathy, not merely a biomarker.[46]
Etiology-Specific Notes
- Asparaginase: Therapeutic asparagine depletion can increase ammonia generation; risk is context dependent.[47] Mitigation aims to preserve anticancer efficacy while limiting toxicity.[48]
- Research insights: Multi-omics studies demonstrate broad alterations in neurotransmission, oxidative-stress responses, mitochondrial function, and protein-synthesis pathways; hypomorphic CPS1 models show disrupted neuronal maturation/synaptic signaling with long-term neurodevelopmental deficits, mirroring clinical observations.[49]
History and Physical
History
The presentation ranges from subtle neurocognitive changes to fulminant encephalopathy, and the age of onset (neonatal, childhood, adult) often reflects the etiology.
Neonatal/early-onset
- Timing: 24 to 72 hours after birth, as nitrogen load rises; ammonia is typically higher than 100 to 150 μmol/L
- Features: Poor feeding, somnolence, irritability, vomiting, hypothermia, hypotonia
- Progression: Hyperventilation with respiratory alkalosis, which can lead to apnea, abnormal posturing, seizures, and coma (≈50% of severe cases develop seizures without rapid therapy) [11]
Late-onset (childhood/adult)
- Contexts: Partial UCDs or acquired causes.
- Precipitants: Infection (eg, gastroenteritis, spontaneous bacterial peritonitis), surgery/trauma, gastrointestinal bleeding, high-protein intake, pregnancy/postpartum, constipation, diuresis, total parenteral nutrition [50]
- Symptoms: Irritability, headache, vomiting, ataxia, confusion/mood change/psychosis; risk of seizures, cerebral edema, and coma increases when ammonia >200 μmol/L
- Protein aversion and cyclical unexplained nausea/vomiting may be present.[51]
- Psychiatric presentations: Consider metabolic evaluation in atypical or refractory cases.[52]
Chronic/recurrent hyperammonemia
- Effects: Intellectual disability, executive dysfunction, learning, and psychiatric disorders
- Mechanism: Linked to astrocytic glutamine accumulation and neurotransmission abnormalities.
- Cirrhosis/minimal hepatic encephalopathy: Fluctuating disorientation, poor concentration, sleep–wake inversion.
- Even mild elevations impair cognition, supporting surveillance and treatment.
Physical Examination
Findings reflect neurologic, systemic, and hepatic involvement and are often nonspecific.
Neurologic
- Early: Impaired attention, irritability, disorientation, slurred speech
- Progressive: Asterixis, ataxia, seizures, altered sensorium → stupor/coma
- Focal deficits: Uncommon in isolated hyperammonemia; prompt evaluation for alternative/additional diagnoses (eg, stroke, central nervous system infection)
- Clinical correlation: Level of consciousness typically worsens as ammonia rises, especially when greater than 200 μmol/L—though correlation is imperfect.[52]
Metabolic clues (inborn errors of metabolism): Isovaleric acidemia (sweaty-feet odor); argininosuccinic aciduria (brittle, fragile hair)
Systemic/hepatic
The stigmata of chronic liver disease and portal hypertension include jaundice, hepatomegaly, ascites with shifting dullness, spider angiomata, gynecomastia, pedal edema, and caput medusae. Fetor hepaticus supports the diagnosis of hepatic encephalopathy. Dehydration and tachypnea may accompany recurrent vomiting and respiratory alkalosis.[53]
Hepatic Encephalopathy Grading (West Haven)
- Grade 0 (minimal): Subclinical; psychometric deficits (attention, memory)
- Grade 1: Sleep–wake reversal, reduced attention, mild disorientation, mood/behavior change
- Grade 2: Lethargy, apathy, disorientation, slurred speech, personality change, asterixis
- Grade 3: Somnolent but arousable; marked confusion, gross disorientation
- Grade 4: Coma with absent/minimal response to pain
Pediatric Considerations
In children, especially with acute liver failure, hepatic encephalopathy presents variably and carries a higher risk of cerebral edema. Maintain a low threshold for intensive care unit-level monitoring and neuroprotective measures when signs of intracranial hypertension emerge.[51]
Evaluation
Newborn Screening
Many disorders (eg, ASS1, ASL, and several organic acidemias) are detected; however, OTC and NAGS are typically not on this screen. A high suspicion despite a normal screen must be maintained.[54][55] Hyperammonemia, higher than 100 μmol/L in neonates and higher than 50 μmol/L in children/adults, is a medical emergency due to the rapid risk of cerebral edema and irreversible injury. Plasma ammonia in unexplained encephalopathy, altered mental status, or seizures should be checked.[56]
Preanalytical essentials
Free-flowing venous/arterial sample in ammonia-free heparin/ethylenediaminetetraacetic acid should be put on ice immediately. Hemolysis and delays should be avoided. Timing and dialysis/procedures should be documented; pre-dialysis samples should be drawn when possible.[57]
Stepwise approach
- A properly handled specimen should be confirmed and repeated if discordant.
- Etiology should be classified as hepatic (acute liver failure, cirrhosis, shunts) or nonhepatic (UCDs/other inborn error of metabolism, drugs, urease-producing infections, nutritional/metabolic causes).
- Initial labs: Comprehensive metabolic panel, liver tests (aspartate aminotransferase/alanine aminotransferase, bilirubin, International Normalized Ratio), arterial blood gas/venous blood gas, glucose, lactate, complete blood count, urinalysis.
- Infectious work-up as indicated (blood/urine cultures; targeted tests) should be performed.
- Metabolic testing should be done if non-hepatic causes are likely: plasma amino acids, urine organic acids, acylcarnitine, urinary orotic acid (elevated in OTC; low/normal in proximal defects). Review newborn screening in neonates (note: OTC/NAGS usually not detected). Consider enzyme assays if genetics are inconclusive.
Imaging and neurophysiology
Brain MRI (edema, chronic injury; UCD patterns), electroencephalogram for seizures/nonconvulsive seizures, and abdominal vascular imaging (Doppler/computed tomography/MRI) for shunts or thrombosis should be performed.[58]
Adult-focused considerations
Partial/late-onset UCDs, urease-producing infections, medication causes, and nutritional deficits should be screened for.[3]
Pitfalls:
- Normal ammonia does not exclude hepatic encephalopathy.
- In cirrhosis, ammonia correlates poorly with grade; very high levels still signal risk.[59][60]
- Intensive care unit underrecognition: In unexplained encephalopathy, ammonia should be checked; involve nephrology/metabolics early.
- Post-dialysis samples can mask elevations—pre-dialysis draws are preferred.
- Seizures (± valproate) can transiently raise ammonia—interpret in context.[61]
- Risk of hyperammonemia increases with valproate, along with topiramate, carnitine deficiency, liver dysfunction, and high drug levels; improves with withdrawal ± carnitine.[3]
- Other culprits, such as asparaginase, chemotherapeutics, antibiotics, and psychotropics, should stop early; consider nitrogen scavengers when indicated.
Clinical insight
Stabilization and ammonia-lowering therapy must begin in parallel with cause identification to prevent irreversible neurologic injury.
Treatment / Management
Treatment of hyperammonemia is time-critical. Stabilization, rapid ammonia reduction, and etiologic testing must proceed in parallel. Core steps are consistent across causes, with disease-specific modifications (UCDs/inborn error of metabolism (IEMs) vs hepatic failure vs drug/infection-related).[62]
Initial stabilization (minutes–hours)
- Airway, breathing, circulation: Secure airway; maintain hemodynamics; admit to intensive care unit (ICU) for severe encephalopathy or suspected cerebral edema.[62]
- Immediate protein restriction: Stop all exogenous protein/nitrogen (including total parenteral nutrition).
- Anti-catabolic calories: Start intravenous (IV) glucose ~8–10 mg/kg/min; add IV lipid as needed to reach ~80–100 kcal/kg/day.[56]
- Correct derangements: Treat hypoglycemia, acidosis, and electrolyte abnormalities; maintain normothermia/euvolemia; use isotonic fluids for hypotension.
- Monitoring: Check ammonia every 2–4 h; perform serial neurological exams. Obtain neuroimaging if edema is suspected; consider intracranial pressure monitoring in fulminant hepatic failure (where available).[62]
- Seizures: Avoid valproate; use levetiracetam or benzodiazepines.
Extracorporeal ammonia removal
- Intermittent hemodialysis (IHD): First-line for severe/refractory hyperammonemia (eg, >300–500 μmol/L, coma, or cerebral edema); clearance superior to PD.[63]
- Continuous renal replacement therapy (CRRT) (continuous venovenous hemodiafiltration): Preferred in neonates or hemodynamic instability; recommended in acute liver failure (ALF).[62]
- Peritoneal dialysis: Less efficient; use only if IHD/CRRT unavailable.
- High-volume plasma exchange: ALF with grade II hepatic encephalopathy or higher may improve transplant-free survival; consider it alongside transplant evaluation.[64]
- ALF emphasis: Hyperammonemia drives cerebral edema and mortality—prioritize early ICU transfer, close ammonia monitoring, and timely RRT.[62]
(B3)
Pharmacologic ammonia-lowering strategies
- Nitrogen scavengers
- IV sodium phenylacetate + sodium benzoate: First-line in acute UCD crises (weight-based loading then maintenance).[56]
- Chronic therapy: oral sodium phenylbutyrate or glycerol phenylbutyrate (often better tolerated).
- Severe non-UCD hyperammonemia: IV scavengers may be considered when a metabolic mechanism is suspected.[56]
- Amino-acid supplementation
- Citrulline: For proximal defects (CPS1, OTC)
- Arginine: For most UCDs except arginase deficiency
- Carglumic acid (N-carbamyl-L-glutamate)
- Indication: NAGS deficiency
- Off-label rescue: Refractory valproate-associated hyperammonemia and selected CPS1 regulatory defects (evidence largely case-based)
- Adjuncts
- L-carnitine: Standard in valproate toxicity; may reduce attack frequency in selected metabolic disorders.
- L-ornithine–L-aspartate, LOLA: Enhances extrahepatic ammonia disposal; effective in hepatic encephalopathy in meta-analyses, though guideline uptake varies.[65]
(A1)
Hepatic encephalopathy–focused care
- First-line: Lactulose/lactitol titrated to 2 to 3 soft stools/day.[65]
- Add-on: rifaximin to reduce recurrence/persistence.[1]
- Selective adjuncts: If response is inadequate, Neomycin or metronidazole (toxicity limits), probiotics/synbiotics, and LOLA.[1]
- Precipitant control: Treat gastrointestinal bleeding, infection, dehydration/constipation, renal failure, and drug toxicity.[1]
- ALF nuance: In ALF, nonabsorbable disaccharides/antibiotics have not shown outcome benefit; prioritize rapid ammonia clearance and definitive therapy. (A1)
Complications and ICU measures
- Cerebral edema/intracranial hypertension: Prefer hypertonic saline over mannitol; elevate head; maintain normocapnia and oxygenation. In fulminant hepatic failure, propofol sedation and controlled ventilation are considered.[66]
- Medication cautions: Avoid valproate; minimize sedatives. Corticosteroids worsen nitrogen balance and are not indicated for hyperammonemia itself.
- Sepsis: Treat aggressively with early source control and antibiotics in parallel with ammonia-lowering therapy. (A1)
Nutrition and long-term management
- Protein strategy: Restrict only during the acute crisis. In chronic liver disease, avoid prolonged restriction; ensure adequate protein to prevent sarcopenia. Limit restriction beyond the acute phase (typically ≤24–48 h) unless guided by metabolic specialists.[1]
- UCD maintenance: Tailored protein-restricted diet ± specialized formulas; oral scavengers; arginine/citrulline and carnitine as indicated.[56]
- Definitive therapies:
- Liver transplantation—curative for many UCDs and for advanced liver failure–associated hyperammonemia.[56]
- Emerging therapies: Gene and messenger ribonucleic acid-based approaches under active investigation.
Special populations
- Very-low-birth-weight neonates: CRRT is feasible/safe for ammonia clearance.
- Pregnancy (UCD carriers/known UCD): Preconception counseling; peripartum plans; aggressive anti-catabolic protocols; rapid access to scavengers/RRT.[56]
- Post-transplant/immunocompromised: Lung-transplant hyperammonemia (often ureaplasma/mycoplasma) carries high mortality without rapid recognition—start targeted antibiotics and timely RRT; standardized screening practices vary.
- Vascular anomalies: Intrahepatic portosystemic shunts; closure or resection can resolve encephalopathy in selected cases.
- Transplant-related: Post-lung or HSCT hyperammonemia (often ureaplasma); requires rapid antimicrobials plus aggressive ammonia-lowering strategies.[67]
Practical pearls
- Treat ammonia when 200 μmol/L or higher (or ≥3× age-adjusted normal in neonates) as a medical emergency.
- Start anticatabolic calories and ammonia-lowering therapy immediately—do not wait for the full workup.
- If levels do not fall within hours on scavengers/support, escalate to RRT.[62]
- Optimal outcomes require early ICU transfer, tight monitoring, and coordinated interprofessional management (hepatology, genetics, nephrology, neurology, nutrition, critical care).[62]
Differential Diagnosis
Hyperammonemia mimics many encephalopathic states. Distinguish true elevation from look-alikes with normal ammonia to avoid treatment delay; evaluate sepsis and metabolic causes in neonates in parallel. In cirrhosis, encephalopathy out of proportion to liver dysfunction warrants a search for precipitants/alternatives.[11][68]
- Infectious and neurologic mimics
- Neonates: Sepsis, meningitis, intracranial hemorrhage (ICH), hypoxic-ischemic encephalopathy—obtain ammonia at triage while starting sepsis care.[11] Adults: Meningitis/encephalitis, stroke/ICH, tumors, posterior reversible encephalopathy syndrome, and idiopathic intracranial hypertension.[68][69] True trigger: Urease-producing urinary tract infection (UTI) (eg, C urealyticum) → true hyperammonemia; treat with targeted antibiotics, urinary decompression, and ammonia-lowering therapy.[70]
- Metabolic disorders with normal ammonia
- Carbohydrate disorders (hypoglycemia), organic acidemias (acidosis/ketosis), pyruvate/mitochondrial disease (lactic acidosis), and homocystinuria (thromboembolism). Uremia usually shows normal/modest ↑ ammonia.
- Secondary mimics/comorbidities
- Post-ictal states, hypoxic–ischemic injury, drug-induced encephalopathy (sedatives/opioids/antiepileptics like valproate may also cause true hyperammonemia), severe sepsis/multiple organ failure, fluoropyrimidines; improve with withdrawal/source control.[71]
- Radiologic overlap
- MRI findings (eg, insular/cingulate diffusion restriction) are nonspecific; magnetic resonance spectrography/diffusion tensor imaging may support them but is not diagnostic.
- Do not miss
Unlike urea cycle disorders, which typically present with hyperammonemia, normal anion gap, and normal glucose, organic acidemias and related metabolic defects are usually characterized by metabolic acidosis with an elevated anion gap, a key distinction in the differential diagnosis.[https://www.ncbi.nlm.nih.gov/books/NBK448090/]
Prognosis
Outcomes depend on etiology, peak ammonia, exposure duration, and speed of intervention. Severe or prolonged elevations lead to cerebral edema, seizures, coma, irreversible injury, and death.[35][62]
Urea Cycle Disorders
- Neonatal-onset: ~35% survival at ~11 years; improving with earlier detection, rapid scavengers, and transplant access.
- Late-onset: ~87% survival at ~11 years with timely recognition/treatment.[6]
- Morbidity: Common neurocognitive/psychiatric sequelae; severity correlates with age at first crisis, peak ammonia, and time to therapy.[6]
- Transplantation: Orthotopic liver transplantation is metabolically curative for many; pediatrics have >85% 10-year survival chance; earlier listing improves neurodevelopment.[74]
Hepatic Encephalopathy
- ALF: Short-term mortality ~50%–70%; hyperammonemia predicts intracranial hypertension and poor survival; urgent transplant evaluation is essential.[62]
- [62]Cirrhosis: Recurrent hepatic encephaly (HE) marks advanced disease; historical 1- and 3-year survival ~42% and 23%; modern outcomes vary by the model for end-stage liver disease (MELD) and transplant access.
Secondary/Drug-Related
This is generally favorable if promptly recognized and the trigger removed (eg, stop valproate, treat infection); prognosis reflects timeliness and baseline organ reserve.[75]
Poor Prognostic Factors
A delay in treatment longer than 24 hours, ammonia levels exceeding 200–300 µmol/L or persistently elevated, the presence of seizures or cerebral edema, coma at presentation, delayed initiation of renal replacement therapy when indicated, and advanced hepatic failure with a high MELD score are critical factors associated with poor outcomes in hyperammonemic conditions such as urea cycle disorders or hepatic encephalopathy.[62][35]
Emerging and Post-Transplant
Biomarkers (plasma glutamine, magnetic resonance spectroscopy of glutamine and glutamate ratios, diffusion metrics) and earlier use of modern scavengers/RRT/timely transplant may improve outcomes. After a transplant for metabolic disease, survival/metabolic stability improves, though neurodevelopment remains variable; early rehabilitation is important.[43][62][76][62][77]
Complications
Hyperammonemia causes acute, life-threatening events and chronic sequelae via direct neurotoxicity, systemic effects of the underlying disease, and treatment-related adverse events.
Acute neurologic complications
- Cerebral edema/intracranial hypertension: Astrocytic glutamine–mediated swelling; rapid progression to herniation without prompt therapy.[62]
- Seizures/status epilepticus: N-methyl-D-aspartate receptor overactivation and energy failure; exacerbate edema and neurologic decline.[46]
- Coma: Severe levels more than 200 to 300 μmol/L in adults often necessitate ICU care; more than 350–400 μmol/L frequently associate with irreversible injury.[46]
- Acute liver failure encephalopathy: Ammonia is a principal driver of asterixis, edema, and progressive coma.[62]
Chronic neurologic sequelae
- Neurocognitive impairment: Developmental/intellectual disability, executive and learning deficits, psychiatric symptoms.[78]
- Motor deficits: Corticospinal-tract injury (advanced imaging) with spasticity and gait disorder.
- Behavioral/psychiatric disorders: Prominent in late-onset UCDs and chronic HE.[78]
Treatment-related complications
- Rapid correction: Rare demyelination with abrupt osmolar shifts; prioritize ammonia reduction while avoiding large sodium/osmolar changes.
- RRT events: Intermittent hemodialysis can cause hypotension, arrhythmias, and disequilibrium; CRRT is better tolerated in instability.
- Nitrogen scavengers: Gastrointestinal upset, anorexia, electrolyte derangements; IV forms add sodium load (worsen edema/heart failure).
- Medication pitfalls: Valproate may precipitate/worsen hyperammonemia—risk ↑ with topiramate or hepatic dysfunction.
Systemic complications
- Multiorgan dysfunction: Contributions to hepatic failure, renal impairment, and coagulopathy.[62]
- Infection susceptibility: Due to hospitalization, malnutrition, and immunosuppression (post-transplant/advanced cirrhosis).
- Growth/nutrition (pediatrics): Excessive protein restriction risks failure to thrive and developmental delay.
Long-term and emerging issues
- Psychiatric burden/quality of life: Increased depression/anxiety in UCD/HE survivors.[79]
- Post-transplant: risks of rejection and immunosuppressant toxicity; rare metabolic recurrence.[77][62]
- Neuroimaging predictors: Diffusion tensor imaging (DTI) and magnetic resonance spectroscopy (MRS) (elevated glutamine/glutamate peaks) may aid risk stratification and rehabilitation planning.
Consultations
Core Team Members and Roles
- Internists/hospitalists recognize early, initiate stabilization, and coordinate multidisciplinary care.
- Emergency medicine triage ammonia testing/handling, begin empiric stabilization, activate specialty pathways.[56]
- Hepatology/gastroenterology manages hepatic encephalopathy and cirrhosis-related hyperammonemia; assesses transplant eligibility.[16]
- Genetics/metabolic diagnosis IEMs guide targeted therapy (eg, carglumic acid and scavengers) and provide genetic counseling.[16]
- Neonatology/pediatrics lead neonatal/infant care.[11]
- Neurology manages seizures; guides neuroimaging/electroencephalogram; assesses neurocognitive outcomes.
- Critical care oversees ICU care, ICP strategies, ventilatory goals, CRRT/hyperammonemic valproate-induced encephalopathy.[62]
- Nephrology initiates IHD/CRRT; manages renal complications.
- Pharmacy optimizes the dosing of scavengers and HE therapies, flags interactions (eg, valproate and corticosteroids), adjusts for hepatic and renal impairment, and builds electronic health record alerts for high-risk agents.
- Dietetics design nutrition plans balancing protein needs with adequate calories.
- Nursing front-line monitoring, therapy administration, infection prevention, and caregiver education.
- Other specialists (surgery/urology/hematology) provide transplant evaluation, urinary interventions, and hematologic issues as needed.
Deterrence and Patient Education
Prevention hinges on patient/caregiver education, early symptom recognition, and etiology-specific plans coordinated by a multidisciplinary team. For UCDs, provide a sick-day plan and an emergency letter (weight-based dosing, ED instructions, key contacts).[56][6]
General education
- Warning signs: Lethargy, confusion, vomiting, gait change, new mood/behavior symptoms.[56]
- Emergency (UCDs): Seek care at the first symptoms; bring the emergency letter.
- Adherence: continue Nitrogen scavengers, ammonia-lowering agents, and supplements; in cirrhosis, do not stop lactulose/rifaximin without guidance.[16]
Diet and lifestyle
- Calories: Maintain adequate intake, especially during illness, to blunt catabolism.[56]
- Protein: UCDs are individualized, specialist-guided restrictions; unless directed, avoid fasting/ketogenic diets. Cirrhosis—no routine restriction; ensure protein to prevent sarcopenia; avoid alcohol; prevent constipation.[16][1]
- Activity: Moderate exercise; avoid prolonged fasting/overexertion
Special populations
- Children (UCDs): Monitor growth/neurodevelopment; ongoing dietetic support.[6]
- Women of childbearing potential: Preconception genetics; coordinated perinatal plan; avoid valproate.
- Families at risk: Offer carrier testing and prenatal/antenatal options.
- Evidence: UCD pregnancies have high maternal/neonatal risk; intensive preconception and peripartum management is required.[80]
Preventing acquired hyperammonemia
- Medications: Review/avoid ammonia-raising agents (eg, valproate, corticosteroids, ribavirin, select chemotherapies) or monitor closely.
- Infections: Meticulous line/urinary/wound care to limit urease-producing pathogens.[17][19]
- Cirrhosis care: Reinforce lactulose/rifaximin adherence, bowel regularity, judicious PPI use, and early reporting of GI bleeding/infection; avoid unsupervised sedatives/opioids.[1][16][81]
Multidisciplinary support
- Dietitians, genetics, and behavioral health.
- Sick-day plans/rescue kits (UCDs): early carbohydrate loading, ED thresholds, on-call contacts.
Key messages
- Act immediately on early symptoms.
- Never stop ammonia-lowering therapy without advice.
- Maintain calories during illness; follow individualized protein targets.
- Keep an updated medication list and flag ammonia-raising drugs.
- Reintroduce protein within 24 to 48 h after an acute episode as directed.
Pearls and Other Issues
Pearls and other important information include the following:
- Treat immediately: Hyperammonemia is an emergency—start anti-catabolic calories and ammonia-lowering therapy without delay.
- Check correctly: Proper specimen handling (free-flowing sample, on ice, rapid transport) prevents false results that delay care.
- Thresholds to act: Very high levels (≈>200 μmol/L adults; ≥3× normal in neonates) or any coma/edema warrant urgent ICU and dialysis planning.
- Dialysis early: If ammonia does not fall within hours or is over 300–500 μmol/L, escalate to IHD (first-line) or CRRT if it is unstable or neonatal.
- UCD crisis: Give IV nitrogen scavengers promptly; add citrulline/arginine per defect; consider carglumic acid for suspected N-acetylglutamate synthase (NAGS) deficiency.
- Hepatic encephalopathy: Titrate lactulose to 2–3 soft stools/day; add rifaximin for recurrence prevention; reintroduce protein within 24 to 48 h.
- Avoid pitfalls: Avoid using valproate (especially with topiramate); search for urease-producing infections and covert portosystemic shunts.
- Pre-dialysis sampling: Draw ammonia before hemodialysis; post-dialysis values can mask severity.
- Nutrition: Restrict protein only during the acute phase; chronic restriction in cirrhosis worsens sarcopenia and outcomes.
- Disposition: Early transfer to centers with renal replacement therapy and transplant/metabolic expertise improves survival and neurologic recovery.
- Stat pearls
- Toxicity increases with both ammonia level and exposure duration, progressing from astrocyte swelling to cerebral edema, seizures, coma, and death; prevention relies on early recognition, rapid ammonia reduction, prompt trigger control, careful therapy titration, and ongoing neurologic surveillance.
- Check ammonia early in unexplained encephalopathy.[68]
- Confirm true elevation and treat triggers in parallel with ammonia-lowering therapy.
- Keep a broad differential in suspected nonhepatic cases (ICU); early scavengers or RRT can be lifesaving.
- Escalating toxicity: Astrocyte swelling → cerebral edema → seizures/coma; greater than 200 μmol/L (adults) or greater than 1000 μmol/L (neonates) demands urgent lowering.[68]
Enhancing Healthcare Team Outcomes
Hyperammonemia is a metabolic emergency that can rapidly cause cerebral edema, intracranial hypertension, encephalopathy, and irreversible brain injury. Early recognition, rapid ammonia reduction, and seamless interprofessional collaboration are essential to improve survival and long-term neurologic outcomes. Patients with hyperammonemia are at high risk of neurologic damage. Early identification and management of patients with this condition are imperative in reducing morbidity and mortality.
The care of patients with hyperammonemia requires a collaborative approach among healthcare professionals to ensure patient-centered care and improve overall outcomes. Neurologists, gastroenterologists, emergency medicine physicians, critical care physicians, genetecists, advanced practitioners, nurses, pharmacists, and other health professionals involved in the care of these patients should possess the essential clinical skills and knowledge to diagnose and manage hyperammonemia accurately. Dietitians are key to avoiding triggering amino acid consumption, and pharmacists aid in antibiotic stewardship. Social workers help provide support to patients and their families as needed. Patients with liver failure should be referred to a transplant network when appropriate. This includes expertise in recognizing the varied clinical presentations and understanding the nuances of diagnostic techniques, such as interpreting lab values and clinical findings.
A strategic approach is equally crucial, involving evidence-based strategies to optimize treatment plans and minimize adverse effects. Ethical considerations must guide decision-making, ensuring informed consent and respecting patient autonomy in treatment choices. Each healthcare professional must know their responsibilities and contribute their unique expertise to the patient's care plan, fostering a multidisciplinary approach. Effective interprofessional communication is paramount, allowing seamless information exchange and collaborative decision-making among the team members. Care coordination plays a pivotal role in ensuring that the patient's journey from diagnosis to treatment and follow-up is well-managed, minimizing errors and enhancing patient safety. By embracing these principles of skill, strategy, ethics, responsibilities, interprofessional communication, and care coordination, healthcare professionals can deliver patient-centered care, ultimately improving patient outcomes and enhancing team performance in the management of hyperammonemia.
Media
(Click Image to Enlarge)
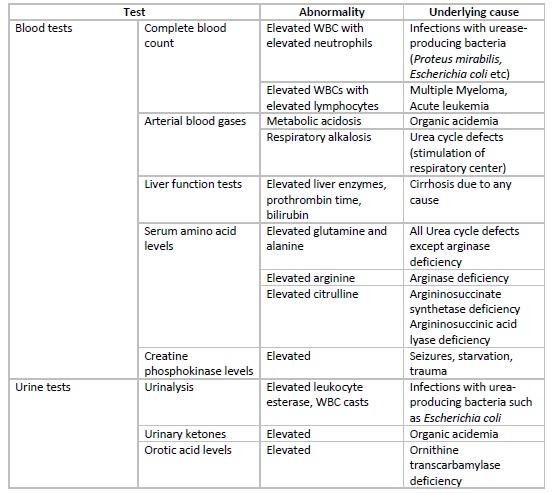
Hyperammonemia, Laboratory Diagnosis Table. This table shows the test, abnormality, and underlying cause of hyperammonemia.
Created and contributed by R Ali, MBBS
(Click Image to Enlarge)
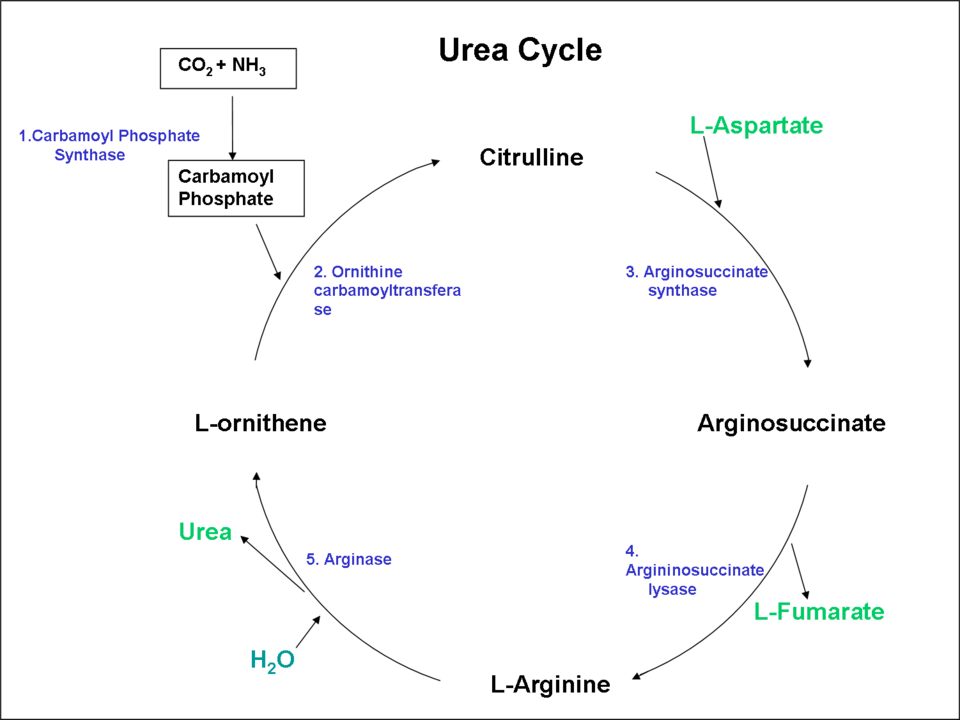
Urea Cycle. This image shows a diagram of the urea cycle pathway.
Eswiss, Public Domain, via Wikipedia
{kind=link}
References
Lauridsen MM, Bajaj JS. Inpatient management of hepatic encephalopathy. Clinical liver disease. 2024 Jan-Jun:23(1):e0105. doi: 10.1097/CLD.0000000000000105. Epub 2024 Feb 4 [PubMed PMID: 38312993]
Meier C, Burns K, Manolikos C, Fatovich D, Bell DA. Hyperammonaemia: review of the pathophysiology, aetiology and investigation. Pathology. 2024 Oct:56(6):763-772. doi: 10.1016/j.pathol.2024.06.002. Epub 2024 Jul 25 [PubMed PMID: 39127541]
Shakerdi L, Ryan A. Drug-induced hyperammonaemia. Journal of clinical pathology. 2023 Aug:76(8):501-509. doi: 10.1136/jcp-2022-208644. Epub 2023 May 10 [PubMed PMID: 37164630]
Martín-Hernández E, Bellusci M, Pérez-Mohand P, Correcher Medina P, Blasco-Alonso J, Morais-López A, de Las Heras J, Meavilla Olivas SM, Dougherty-de Miguel L, Couce ML, Villarroya EC, García Jiménez MC, Moreno-Lozano PJ, Vives I, Gil-Campos M, Stanescu S, Ceberio-Hualde L, Camprodón M, Cortès-Saladelafont E, López-Urdiales R, Murray Hurtado M, Márquez Armenteros AM, Sierra Córcoles C, Peña-Quintana L, Ruiz-Pons M, Alcalde C, Castellanos-Pinedo F, Dios E, Barrio-Carreras D, Martín-Cazaña M, García-Peris M, Andrade JD, García-Volpe C, de Los Santos M, García-Cazorla A, Del Toro M, Felipe-Rucián A, Comino Monroy MJ, Sánchez-Pintos P, Matas A, Gil Ortega D, Martín-Rivada Á, Bergua A, Belanger-Quintana A, Vitoria I, Yahyaoui R, Pérez B, Morales-Conejo M, Quijada-Fraile P. Understanding the Natural History and the Effects of Current Therapeutic Strategies on Urea Cycle Disorders: Insights from the UCD Spanish Registry. Nutrients. 2025 Mar 28:17(7):. doi: 10.3390/nu17071173. Epub 2025 Mar 28 [PubMed PMID: 40218931]
Level 3 (low-level) evidenceGemici Karaaslan B, Kiykim A, Burtecene N, Gokden M, Cansever MS, Hopurcuoglu D, Cengiz GN, Topcu B, Zubarioğlu T, Kiykim E, Cokuğras H, Aktuglu Zeybek AC. Amino Acid Metabolism and Immune Dysfunction in Urea Cycle Disorders: T and B Cell Perspectives. Journal of inherited metabolic disease. 2025 Mar:48(2):e70009. doi: 10.1002/jimd.70009. Epub [PubMed PMID: 39957310]
Level 3 (low-level) evidenceBurlina A, Gasperini S, la Marca G, Pession A, Siri B, Spada M, Ruoppolo M, Tummolo A. Long-Term Management of Patients with Mild Urea Cycle Disorders Identified through the Newborn Screening: An Expert Opinion for Clinical Practice. Nutrients. 2023 Dec 20:16(1):. doi: 10.3390/nu16010013. Epub 2023 Dec 20 [PubMed PMID: 38201843]
Level 3 (low-level) evidenceDas AM. Urea cycle defects in adulthood: clinical presentation, diagnosis and treatment in genetically encoded hepatic metabolic disorders with a potential for encephalopathy. Metabolic brain disease. 2025 Apr 26:40(5):192. doi: 10.1007/s11011-025-01619-5. Epub 2025 Apr 26 [PubMed PMID: 40285952]
Noori M, Jarrah O, Al Shamsi A. Carbamoly-phosphate synthetase 1 (CPS1) deficiency: A tertiary center retrospective cohort study and literature review. Molecular genetics and metabolism reports. 2024 Dec:41():101156. doi: 10.1016/j.ymgmr.2024.101156. Epub 2024 Oct 18 [PubMed PMID: 39469307]
Level 2 (mid-level) evidenceBin Sawad A, Pothukuchy A, Badeaux M, Hodson V, Bubb G, Lindsley K, Uyei J, Diaz GA. Natural history of arginase 1 deficiency and the unmet needs of patients: A systematic review of case reports. JIMD reports. 2022 Jul:63(4):330-340. doi: 10.1002/jmd2.12283. Epub 2022 Mar 25 [PubMed PMID: 35822089]
Level 1 (high-level) evidenceDiaz GA, Bechter M, Cederbaum SD. The role and control of arginine levels in arginase 1 deficiency. Journal of inherited metabolic disease. 2023 Jan:46(1):3-14. doi: 10.1002/jimd.12564. Epub 2022 Oct 13 [PubMed PMID: 36175366]
Ni B, Qin M, Zhao J, Guo Q. A glance at transient hyperammonemia of the newborn: Pathophysiology, diagnosis, and treatment: A review. Medicine. 2022 Dec 2:101(48):e31796. doi: 10.1097/MD.0000000000031796. Epub [PubMed PMID: 36482558]
Level 2 (mid-level) evidenceRao NN, Burns K, Manolikos C, Hodge S. Late-onset multiple acyl-CoA dehydrogenase deficiency: an insidious presentation. BMJ case reports. 2023 May 22:16(5):. doi: 10.1136/bcr-2022-252668. Epub 2023 May 22 [PubMed PMID: 37217231]
Level 3 (low-level) evidenceAdam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Song YZ, Oishi K, Saheki T. Citrin Deficiency. GeneReviews(®). 1993:(): [PubMed PMID: 20301360]
Kirchberg I, Lainka E, Gangfuß A, Kuechler A, Baertling F, Schlieben LD, Lenz D, Tschiedel E. Distinct neonatal hyperammonemia and liver synthesis dysfunction: case report of a severe MEGDHEL syndrome. Frontiers in pediatrics. 2024:12():1278047. doi: 10.3389/fped.2024.1278047. Epub 2024 Feb 20 [PubMed PMID: 38445077]
Level 3 (low-level) evidenceWalti LN, Ng CF, Kaur S, Almansour S, Mazzulli T, Bitterman R, Sidhu A, Keshavjee S, Chaparro C, Martinu T, Tikkanen J, Del Sorbo L, Husain S. Serum Ammonia Screening and Donor Mollicutes Detection for Hyperammonemia Syndrome Post-Lung Transplantation: A Prospective Observational Study. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2025 Dec 24:81(5):998-1004. doi: 10.1093/cid/ciaf078. Epub [PubMed PMID: 39985813]
Level 2 (mid-level) evidenceEuropean Association for the Study of the Liver. EASL Clinical Practice Guidelines on the management of hepatic encephalopathy. Journal of hepatology. 2022 Sep:77(3):807-824. doi: 10.1016/j.jhep.2022.06.001. Epub 2022 Jun 17 [PubMed PMID: 35724930]
Level 1 (high-level) evidenceNishioka H, Kusu R. Hyperammonemia caused by urinary tract infection due to Corynebacteriumurealyticum. Journal of infection and chemotherapy : official journal of the Japan Society of Chemotherapy. 2025 May:31(5):102688. doi: 10.1016/j.jiac.2025.102688. Epub 2025 Mar 27 [PubMed PMID: 40157572]
Roberts SC, Bharat A, Kurihara C, Tomic R, Ison MG. Impact of Screening and Treatment of Ureaplasma species on Hyperammonemia Syndrome in Lung Transplant Recipients: A Single Center Experience. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2021 Nov 2:73(9):e2531-e2537. doi: 10.1093/cid/ciaa1570. Epub [PubMed PMID: 33068392]
Nakamura A, Odo M. Hyperammonemia and Impaired Consciousness Caused by Non-Urease-Producing Actinotignum schaalii in Obstructive Urinary Tract Infection. Cureus. 2024 Dec:16(12):e76167. doi: 10.7759/cureus.76167. Epub 2024 Dec 21 [PubMed PMID: 39840191]
Hapshy V, Imburgio S, Sanekommu H, Nightingale B, Taj S, Hossain MA, Patel S. Pylephlebitis-induced acute liver failure: A case report and review of literature. World journal of hepatology. 2024 Jan 27:16(1):103-108. doi: 10.4254/wjh.v16.i1.103. Epub [PubMed PMID: 38313245]
Level 3 (low-level) evidenceSammar A, Tawfik M, Fatima F, Butler A, Aylor-Lee K. Valproate-Induced Hyperammonemic Encephalopathy Causing New-Onset Seizures. Cureus. 2023 Oct:15(10):e47288. doi: 10.7759/cureus.47288. Epub 2023 Oct 18 [PubMed PMID: 38021840]
Kumar A, Advani S, Asim K, Mohamed MA, Wani F, Singh J, Albosta M, Shiwalkar N, Keshavamurthy S. Hyperammonemia in lung transplant patients and its management: a review. Indian journal of thoracic and cardiovascular surgery. 2022 Jul:38(Suppl 2):335-346. doi: 10.1007/s12055-021-01319-6. Epub 2022 Mar 14 [PubMed PMID: 35756952]
Inoue S, Yasuda H, Yoshida K, Mori K, Ogawa K, Yokotsuka Y, Okamoto H. A Diabetic Patient with Prolonged Hyperammonemia Due to Urinary Tract Infection Caused by Urease-producing Bacteria. Internal medicine (Tokyo, Japan). 2024 Jul 1:63(13):1945-1949. doi: 10.2169/internalmedicine.2817-23. Epub 2023 Nov 6 [PubMed PMID: 37926530]
Balcerac A, Bihan K, Lebrun-Vignes B, Thabut D, Salem JE, Weiss N. Drug-associated hyperammonaemia: a Bayesian analysis of the WHO Pharmacovigilance Database. Annals of intensive care. 2022 Jun 18:12(1):55. doi: 10.1186/s13613-022-01026-4. Epub 2022 Jun 18 [PubMed PMID: 35716335]
Vaidyanathan S, Chatorikar S, Praharaj SK, Munoli RN, Udupa ST. Valproate-Associated Hyperammonemic Encephalopathy: Clinical Correlates and Management Strategies in a Tertiary Care Center. Journal of clinical psychopharmacology. 2023 Mar-Apr 01:43(2):145-148. doi: 10.1097/JCP.0000000000001659. Epub 2023 Feb 16 [PubMed PMID: 36795014]
Barnhill M, Yaman R, Renszel K, Alzubaidi S, Vargas H, Chascsa D. Not Your Typical Hyperammonemia: Non-hepatic Severe Hyperammonemia in an Adult. ACG case reports journal. 2023 Dec:10(12):e01225. doi: 10.14309/crj.0000000000001225. Epub 2023 Dec 13 [PubMed PMID: 38093784]
Level 3 (low-level) evidenceDonovan K, Vaqar S, Guzman N. Ornithine Transcarbamylase Deficiency. StatPearls. 2026 Jan:(): [PubMed PMID: 30725942]
Therrell BL, Padilla CD, Borrajo GJC, Khneisser I, Schielen PCJI, Knight-Madden J, Malherbe HL, Kase M. Current Status of Newborn Bloodspot Screening Worldwide 2024: A Comprehensive Review of Recent Activities (2020-2023). International journal of neonatal screening. 2024 May 23:10(2):. doi: 10.3390/ijns10020038. Epub 2024 May 23 [PubMed PMID: 38920845]
Bisht S, Morales A, Jaishankar GB. Urea Cycle Disorders. StatPearls. 2026 Jan:(): [PubMed PMID: 29493985]
Feigenbaum A, Lamale-Smith L, Weinstein L. Considerations for prenatal and postpartum management of a female patient with ornithine transcarbamylase deficiency. Molecular genetics and metabolism reports. 2022 Dec:33(Suppl 1):100894. doi: 10.1016/j.ymgmr.2022.100894. Epub 2022 Jul 12 [PubMed PMID: 36620386]
Kjærgaard K, Eriksen PL, Nøhr TK, Pedersen SB, Gravholt CH, Vilstrup H, Thomsen KL. Hyperammonaemic encephalopathy due to non-functioning urea cycle as a complication to gastric bypass surgery. Metabolic brain disease. 2024 Nov 28:40(1):46. doi: 10.1007/s11011-024-01434-4. Epub 2024 Nov 28 [PubMed PMID: 39607664]
Lu K. Cellular Pathogenesis of Hepatic Encephalopathy: An Update. Biomolecules. 2023 Feb 19:13(2):. doi: 10.3390/biom13020396. Epub 2023 Feb 19 [PubMed PMID: 36830765]
Di Cola S, Nardelli S, Ridola L, Gioia S, Riggio O, Merli M. Ammonia and the Muscle: An Emerging Point of View on Hepatic Encephalopathy. Journal of clinical medicine. 2022 Jan 26:11(3):. doi: 10.3390/jcm11030611. Epub 2022 Jan 26 [PubMed PMID: 35160063]
Buckholz AP, Brown RS Jr. Future Therapies of Hepatic Encephalopathy. Clinics in liver disease. 2024 May:28(2):331-344. doi: 10.1016/j.cld.2024.02.002. Epub [PubMed PMID: 38548443]
Ott P, Eriksen PL, Kjærgaard K, Sørensen M, Thomsen KL, Vilstrup H. Down the road towards hepatic encephalopathy. The elusive ammonia- what determines the arterial concentration? Metabolic brain disease. 2024 Dec 2:40(1):48. doi: 10.1007/s11011-024-01435-3. Epub 2024 Dec 2 [PubMed PMID: 39621139]
Chandel NS. Amino Acid Metabolism. Cold Spring Harbor perspectives in biology. 2021 Apr 1:13(4):. doi: 10.1101/cshperspect.a040584. Epub 2021 Apr 1 [PubMed PMID: 33795250]
Level 3 (low-level) evidenceKosenko E, Tikhonova L, Alilova G, Montoliu C. Is NMDA-Receptor-Mediated Oxidative Stress in Mitochondria of Peripheral Tissues the Essential Factor in the Pathogenesis of Hepatic Encephalopathy? Journal of clinical medicine. 2022 Feb 4:11(3):. doi: 10.3390/jcm11030827. Epub 2022 Feb 4 [PubMed PMID: 35160278]
Li M, Liu Z, Lai K, Liu H, Gong L, Shi H, Zhang W, Wang H, Shi H. Enhanced recruitment of glutamate receptors underlies excitotoxicity of mitral cells in acute hyperammonemia. Frontiers in cellular neuroscience. 2022:16():1002671. doi: 10.3389/fncel.2022.1002671. Epub 2022 Oct 28 [PubMed PMID: 36385944]
Sepehrinezhad A, Shahbazi A, Sahab Negah S, Stolze Larsen F. New Insight Into Mechanisms of Hepatic Encephalopathy: An Integrative Analysis Approach to Identify Molecular Markers and Therapeutic Targets. Bioinformatics and biology insights. 2023:17():11779322231155068. doi: 10.1177/11779322231155068. Epub 2023 Feb 17 [PubMed PMID: 36814683]
Llansola M, Arenas YM, Sancho-Alonso M, Mincheva G, Palomares-Rodriguez A, Doverskog M, Izquierdo-Altarejos P, Felipo V. Neuroinflammation alters GABAergic neurotransmission in hyperammonemia and hepatic encephalopathy, leading to motor incoordination. Mechanisms and therapeutic implications. Frontiers in pharmacology. 2024:15():1358323. doi: 10.3389/fphar.2024.1358323. Epub 2024 Mar 15 [PubMed PMID: 38560359]
Sørensen M, Andersen JV, Bjerring PN, Vilstrup H. Hepatic encephalopathy as a result of ammonia-induced increase in GABAergic tone with secondary reduced brain energy metabolism. Metabolic brain disease. 2024 Nov 19:40(1):19. doi: 10.1007/s11011-024-01473-x. Epub 2024 Nov 19 [PubMed PMID: 39560844]
Yang H, You L, Wang Z, Yang L, Wang X, Wu W, Zhi H, Rong G, Sheng Y, Liu X, Liu L. Bile duct ligation elevates 5-HT levels in cerebral cortex of rats partly due to impairment of brain UGT1A6 expression and activity via ammonia accumulation. Redox biology. 2024 Feb:69():103019. doi: 10.1016/j.redox.2023.103019. Epub 2023 Dec 28 [PubMed PMID: 38163420]
Enokizono M, Aida N, Yagishita A, Nakata Y, Ideguchi R, Kurokawa R, Kono T, Moritani T, Mori H. Neuroimaging findings of inborn errors of metabolism: urea cycle disorders, aminoacidopathies, and organic acidopathies. Japanese journal of radiology. 2023 Jul:41(7):683-702. doi: 10.1007/s11604-023-01396-0. Epub 2023 Feb 2 [PubMed PMID: 36729192]
Thomsen KL, Eriksen PL, Kerbert AJ, De Chiara F, Jalan R, Vilstrup H. Role of ammonia in NAFLD: An unusual suspect. JHEP reports : innovation in hepatology. 2023 Jul:5(7):100780. doi: 10.1016/j.jhepr.2023.100780. Epub 2023 Apr 25 [PubMed PMID: 37425212]
Kerbert AJC, Engelmann C, Habtesion A, Kumar P, Hassan M, Qi T, Volkert I, Otto T, Hall A, Khetan VU, Olde Damink S, Aguilar F, Chollet C, Brunet L, Clària J, Moreau R, Arroyo V, Coenraad MJ, Mehta G, Castelli F, Trautwein C, Fenaille F, Andreola F, Jalan R. Hyperammonemia induces programmed liver cell death. Science advances. 2025 Mar 7:11(10):eado1648. doi: 10.1126/sciadv.ado1648. Epub 2025 Mar 7 [PubMed PMID: 40053595]
Level 2 (mid-level) evidenceAngelova PR, Kerbert AJC, Habtesion A, Hall A, Abramov AY, Jalan R. Hyperammonaemia induces mitochondrial dysfunction and neuronal cell death. JHEP reports : innovation in hepatology. 2022 Aug:4(8):100510. doi: 10.1016/j.jhepr.2022.100510. Epub 2022 May 23 [PubMed PMID: 35845295]
Raja RA, Als-Nielsen B, Lund AM, Vilstrup H, Dalhoff KP, Schmiegelow K. Asparaginase-associated hyperammonemia. Haematologica. 2025 Aug 1:110(8):1702-1709. doi: 10.3324/haematol.2025.287301. Epub 2025 Apr 24 [PubMed PMID: 40270200]
Lee A, Eldem I, Altintas B, Nguyen H, Willis D, Langley R, Shinawi M. Treatment and outcomes of symptomatic hyperammonemia following asparaginase therapy in children with acute lymphoblastic leukemia. Molecular genetics and metabolism. 2023 Jul:139(3):107627. doi: 10.1016/j.ymgme.2023.107627. Epub 2023 Jun 7 [PubMed PMID: 37327713]
Bakshi S, Diep T, Willis BJ, Reyes R, Wu GF, Makris G, Poms M, Day I, Sun Q, Zhuravka I, Lueptow L, Tang M, Cromie GA, Dudley AM, Häberle J, Lipshutz GS. A hypomorphic model of CPS1 deficiency for investigating the effects of hyperammonemia on the developing nervous system. Disease models & mechanisms. 2025 Jul 1:18(7):. doi: 10.1242/dmm.052303. Epub 2025 Jun 20 [PubMed PMID: 40421838]
Ibrahim MS, Gold JI, Woodall A, Yilmaz BS, Gissen P, Stepien KM. Diagnostic and Management Issues in Patients with Late-Onset Ornithine Transcarbamylase Deficiency. Children (Basel, Switzerland). 2023 Aug 9:10(8):. doi: 10.3390/children10081368. Epub 2023 Aug 9 [PubMed PMID: 37628367]
Mandiga P, Kommu S, Bollu PC. Hepatic Encephalopathy. StatPearls. 2026 Jan:(): [PubMed PMID: 28613619]
Mumdzhiev N, Tenev RV, Radicheva MP. Psychometric hepatic encephalopathy score (PHES) - when, how, why, and why not: a guide for the unfamiliar. Przeglad gastroenterologiczny. 2025:20(1):31-35. doi: 10.5114/pg.2024.145382. Epub 2024 Dec 2 [PubMed PMID: 40191504]
Thanapirom K, Wongwandee M, Suksawatamnuay S, Thaimai P, Siripon N, Makhasen W, Treeprasertsuk S, Komolmit P. Psychometric Hepatic Encephalopathy Score for the Diagnosis of Minimal Hepatic Encephalopathy in Thai Cirrhotic Patients. Journal of clinical medicine. 2023 Jan 8:12(2):. doi: 10.3390/jcm12020519. Epub 2023 Jan 8 [PubMed PMID: 36675448]
Tran DM, Tran TTT, Luong QH, Tran MTC. A preliminary retrospective evaluation of screening and diagnosis of ornithine transcarbamylase deficiency in high-risk patients at a referral center in Vietnam. Heliyon. 2024 Aug 30:10(16):e36003. doi: 10.1016/j.heliyon.2024.e36003. Epub 2024 Aug 8 [PubMed PMID: 39220945]
Level 2 (mid-level) evidenceHall PL, Wittenauer AL, Wilcox WR. Proximal urea cycle defects are challenging to detect with newborn screening: Results of a prospective pilot study using post-analytical tools. American journal of medical genetics. Part C, Seminars in medical genetics. 2022 Jun:190(2):178-186. doi: 10.1002/ajmg.c.31996. Epub 2022 Sep 13 [PubMed PMID: 36097743]
Level 3 (low-level) evidenceBélanger-Quintana A, Arrieta Blanco F, Barrio-Carreras D, Bergua Martínez A, Cañedo Villarroya E, García-Silva MT, Lama More R, Martín-Hernández E, López AM, Morales-Conejo M, Pedrón-Giner C, Quijada-Fraile P, Stanescu S, Casanova MM. Recommendations for the Diagnosis and Therapeutic Management of Hyperammonaemia in Paediatric and Adult Patients. Nutrients. 2022 Jul 2:14(13):. doi: 10.3390/nu14132755. Epub 2022 Jul 2 [PubMed PMID: 35807935]
Jeraiby MA. To Investigate the Laboratory Sample Criteria of Ammonia Measurement in Saudi Arabia. Annals of African medicine. 2025 Jul 1:24(3):573-578. doi: 10.4103/aam.aam_80_24. Epub 2025 May 30 [PubMed PMID: 40445302]
Chanvanichtrakool M, Schreiber JM, Chen WL, Barber J, Zhang A, Ah Mew N, Schulze A, Wilkening G, Nagamani SCS, Gropman A, Urea Cycle Disease Consortium. Unraveling the Link: Seizure Characteristics and Ammonia Levels in Urea Cycle Disorder During Hyperammonemic Crises. Pediatric neurology. 2024 Oct:159():48-55. doi: 10.1016/j.pediatrneurol.2024.06.013. Epub 2024 Jun 29 [PubMed PMID: 39121557]
Bajaj JS, Pyrsopoulos NT, Rahimi RS, Heimanson Z, Allen C, Rockey DC. Serum Ammonia Levels Do Not Correlate With Overt Hepatic Encephalopathy Severity in Hospitalized Patients With Cirrhosis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2024 Sep:22(9):1950-1952.e1. doi: 10.1016/j.cgh.2024.02.015. Epub 2024 Feb 28 [PubMed PMID: 38423347]
Deutsch-Link S, Moon AM. The Ongoing Debate of Serum Ammonia Levels in Cirrhosis: the Good, the Bad, and the Ugly. The American journal of gastroenterology. 2023 Jan 1:118(1):10-13. doi: 10.14309/ajg.0000000000001966. Epub 2022 Aug 23 [PubMed PMID: 36001400]
Pan C, Zhao Z, Liu Z, Luo T, Zhu M, Xu Z, Yu C, Huang H. Valproate encephalopathy: Case series and literature review. SAGE open medical case reports. 2024:12():2050313X241260152. doi: 10.1177/2050313X241260152. Epub 2024 Jun 20 [PubMed PMID: 38911175]
Level 2 (mid-level) evidenceTujios S, Stravitz RT, Lee WM. Management of Acute Liver Failure: Update 2022. Seminars in liver disease. 2022 Aug:42(3):362-378. doi: 10.1055/s-0042-1755274. Epub 2022 Aug 24 [PubMed PMID: 36001996]
Raina R, Bedoyan JK, Lichter-Konecki U, Jouvet P, Picca S, Mew NA, Machado MC, Chakraborty R, Vemuganti M, Grewal MK, Bunchman T, Sethi SK, Krishnappa V, McCulloch M, Alhasan K, Bagga A, Basu RK, Schaefer F, Filler G, Warady BA. Consensus guidelines for management of hyperammonaemia in paediatric patients receiving continuous kidney replacement therapy. Nature reviews. Nephrology. 2020 Aug:16(8):471-482. doi: 10.1038/s41581-020-0267-8. Epub 2020 Apr 8 [PubMed PMID: 32269302]
Level 3 (low-level) evidenceGoel A, Zachariah U, Daniel D, Eapen CE. Growing Evidence for Survival Benefit with Plasma Exchange to Treat Liver Failure. Journal of clinical and experimental hepatology. 2023 Nov-Dec:13(6):1061-1073. doi: 10.1016/j.jceh.2023.06.002. Epub 2023 Jun 14 [PubMed PMID: 37975044]
Zhang H, Fu Y, Lin M, Nan Z, Zhao D. Efficacy and safety of L-ornithine L-aspartate combined with lactulose in treatment of hepatic encephalopathy: a systematic review and meta-analysis of randomized controlled trial. Frontiers in medicine. 2025:12():1581792. doi: 10.3389/fmed.2025.1581792. Epub 2025 Apr 30 [PubMed PMID: 40370740]
Level 1 (high-level) evidenceKalal CR, Maiwall R, Choudhary A, Premkumar M, Kumar G, Vyas AK, Sarin SK. Mannitol Is Comparable to Hypertonic Saline for Raised Intracranial Pressure in Acute Liver Failure (MAHAL Study): A Randomized Controlled Trial. Digestive diseases (Basel, Switzerland). 2022:40(5):607-615. doi: 10.1159/000520229. Epub 2021 Nov 11 [PubMed PMID: 34763338]
Level 1 (high-level) evidenceYun S, Scalia C, Farghaly S. Treatment of Hyperammonemia Syndrome in Lung Transplant Recipients. Journal of clinical medicine. 2023 Nov 8:12(22):. doi: 10.3390/jcm12226975. Epub 2023 Nov 8 [PubMed PMID: 38002590]
Bellafante D, Gioia S, Faccioli J, Riggio O, Ridola L, Nardelli S. The Management of Hepatic Encephalopathy from Ward to Domiciliary Care: Current Evidence and Gray Areas. Journal of clinical medicine. 2023 Dec 27:13(1):. doi: 10.3390/jcm13010166. Epub 2023 Dec 27 [PubMed PMID: 38202173]
Zelaya JE, Al-Khoury L. Posterior Reversible Encephalopathy Syndrome. StatPearls. 2026 Jan:(): [PubMed PMID: 32119379]
Ohyama K, Sasaki H, Doi Y, Uehara Y. Encrusted pyelitis and hyperammonemia due to Corynebacterium urealyticum in a kidney transplant recipient. Journal of infection and chemotherapy : official journal of the Japan Society of Chemotherapy. 2025 Feb:31(2):102565. doi: 10.1016/j.jiac.2024.11.015. Epub 2024 Nov 21 [PubMed PMID: 39580053]
Scott A, Rao SV, Affronti ML. Hyperammonemia Secondary to 5-Fluorouracil. Journal of the advanced practitioner in oncology. 2023 Jul:14(5):414-418. doi: 10.6004/jadpro.2023.14.5.6. Epub 2023 Jul 1 [PubMed PMID: 37576363]
Tachibana H, Ohashi N, Okumura G, Tanaka R, Fuseya S, Gotoh S, Ishida T, Shimizu S, Kawamata M, Tanaka S. Postoperative hyperammonemic encephalopathy due to unexpected constipation in a patient with hyperornithinemia-hyperammonemia-homocitrullinuria syndrome: a case report. JA clinical reports. 2024 Jun 21:10(1):42. doi: 10.1186/s40981-024-00726-z. Epub 2024 Jun 21 [PubMed PMID: 38904738]
Level 3 (low-level) evidenceGariani K, Klauser A, Vargas MI, Lazeyras F, Tran C. New Insight in Hyperinsulinism/Hyperammonemia Syndrome by Magnetic Resonance Imaging and Spectroscopy. Brain sciences. 2022 Mar 15:12(3):. doi: 10.3390/brainsci12030389. Epub 2022 Mar 15 [PubMed PMID: 35326344]
García Vega M, Andrade JD, Morais A, Frauca E, Muñoz Bartolo G, Lledín MD, Bergua A, Hierro L. Urea cycle disorders and indications for liver transplantation. Frontiers in pediatrics. 2023:11():1103757. doi: 10.3389/fped.2023.1103757. Epub 2023 Mar 3 [PubMed PMID: 36937980]
Loser V, Novy J, Beuchat I, Rossetti AO. Acute Valproate-Induced Encephalopathy in Status Epilepticus: A Registry-Based Assessment. CNS drugs. 2023 Aug:37(8):725-731. doi: 10.1007/s40263-023-01024-5. Epub 2023 Jul 19 [PubMed PMID: 37466895]
Sen K, Anderson AA, Whitehead MT, Gropman AL. Review of Multi-Modal Imaging in Urea Cycle Disorders: The Old, the New, the Borrowed, and the Blue. Frontiers in neurology. 2021:12():632307. doi: 10.3389/fneur.2021.632307. Epub 2021 Apr 28 [PubMed PMID: 33995244]
Patterson C, Gold A, So S, Kahnami L, Dworsky-Fried M, Mamak E, Rogers A, Schulze A, Ertl-Wagner B, Ng V, Avitzur Y. Long-term neurodevelopmental outcomes following liver transplantation for metabolic disease-a single centre experience. Journal of inherited metabolic disease. 2025 Jan:48(1):e12785. doi: 10.1002/jimd.12785. Epub 2024 Aug 12 [PubMed PMID: 39135350]
Murali CN, Barber JR, McCarter R, Zhang A, Gallant N, Simpson K, Dorrani N, Wilkening GN, Hays RD, Lichter-Konecki U, Members of the Urea Cycle Disorders Consortium, Burrage LC, Nagamani SCS. Health-related quality of life in a systematically assessed cohort of children and adults with urea cycle disorders. Molecular genetics and metabolism. 2023 Nov:140(3):107696. doi: 10.1016/j.ymgme.2023.107696. Epub 2023 Sep 8 [PubMed PMID: 37690181]
Level 1 (high-level) evidenceGarcia-Altieri M, Carrera-Mejias K, Hernaez R. Management of depression/anxiety in patients with chronic liver disease. Clinical liver disease. 2024 Jan-Jun:23(1):e0179. doi: 10.1097/CLD.0000000000000179. Epub 2024 Jun 5 [PubMed PMID: 38855042]
Stepien KM, Langendonk JG, Dao M, Gomes DC, Douillard C, Filipsson K, Glamuzina E, Haverkamp JA, Langeveld M, Lehman A, de Lonlay P, Lund AM, Oscarson M, Peltenburg NC, Ramadža DP, Ramachandran R, Reismann P, Shtylla A, Tchan M, Tan CY, Wilson C, Woodall A, Murphy E, Wagenmakers MAEM. The management and clinical outcomes of pregnancies in women with urea cycle disorders: A review of the literature and results of an international survey. Journal of inherited metabolic disease. 2024 Nov:47(6):1239-1259. doi: 10.1002/jimd.12695. Epub 2023 Dec 9 [PubMed PMID: 38069502]
Level 2 (mid-level) evidenceSanyal AJ, Kowdley KV, Reau NS, Pyrsopoulos NT, Allen C, Heimanson Z, Bajaj JS. Rifaximin plus lactulose versus lactulose alone for reducing the risk of HE recurrence. Hepatology communications. 2024 Jun 1:8(6):. doi: 10.1097/HC9.0000000000000436. Epub 2024 May 10 [PubMed PMID: 38727685]