Introduction
Hirschsprung disease is a congenital disorder characterized by the absence of ganglion cells in the Meissner plexus of the submucosa and the Auerbach plexus of the muscularis in the terminal rectum, which extends a variable distance proximally.[1] In the US, Hirschsprung disease is estimated to affect approximately 1 in every 5000 newborns.[2] Hirschsprung disease remains a significant cause of neonatal intestinal obstruction. Globally, incidence varies by region, with results from studies from the Federated States of Micronesia reporting an incidence approaching 1 in 3000 live births.[3]
Once considered fatal, data from the EUROlinkCAT collaborative network, incorporating 13 population-based congenital anomaly registries, reported a 1-year mortality rate of 2.9% among children with Hirschsprung disease. When extended to 5-year follow-up, cumulative mortality increased modestly to 3.4%.[4] Approximately 80% to 90% of children with Hirschsprung disease become symptomatic in the neonatal period. Presentation after early childhood is uncommon; fewer than 10% of cases are diagnosed after 4 years of age, typically reflecting short-segment disease with milder or partially compensated symptoms.[5] Hirschsprung disease causes nonspecific symptomatology, including chronic constipation and intestinal obstruction.[6] Diagnosis relies on histopathologic examination of rectal biopsies.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
In Hirschsprung disease, the migration and differentiation of neural crest cells within the enteric nervous system are disrupted, a process regulated in part by the rearranged during transfection (RET) gene and its ligands. Enteric neurons are functionally organized into intrinsic primary afferent neurons, which detect luminal distension; ascending and descending interneurons, which transmit sensory signals proximally and distally along the bowel; and motor neurons, which are subdivided into excitatory and inhibitory types that mediate smooth muscle contraction and relaxation, respectively.[7] The disturbance results in a complete absence of ganglion cells in the nerve plexuses. Aganglionosis leads to intestinal overactivity and persistent acetylcholine release. Subsequently, continuous contraction of the narrowed affected colonic segment occurs, with progressive secondary dilatation of the healthy proximal colon.[8]
Hirschsprung disease transmission is complex, involving multigenic inheritance, and the disease commonly presents in a sporadic form (80% to 90%). Penetrance is weak, variable, and sex-dependent. Approximately 10% to 20% of cases of Hirschsprung disease occur in a familial pattern. Familial cases most commonly demonstrate autosomal dominant inheritance with variable penetrance and are more frequently associated with long-segment involvement or total colonic aganglionosis.[9] The main gene involved is the rearranged during transfection (RET) proto-oncogene , found in about 35% of sporadic cases and 49% of familial cases.[10] RET mutations may occur in any of the 21 exons of the gene, and more than 100 different mutations have been identified. Mutations include nonsense, missense, deletions, and insertions.[1][11] The other genes implicated in the etiopathogenesis of Hirschsprung disease account for only 5% to 10% of cases. These genes include the ligands of the RET receptor: glial cell line–derived neurotrophic factor gene (GDNF), endothelin 3 gene (EDN3), endothelin receptor type B gene (EDNRB), SRY-box transcription factor 10 gene (SOX10), and the paired-like homeobox 2B gene (PHOX2B).[1] Hirschsprung disease is associated with several genetic syndromes and chromosomal anomalies. These include Shah–Waardenburg syndrome (EDN3, EDNRB, SOX10), Haddad syndrome (PHOX2B), Mowat–Wilson syndrome (ZEB2), Goldberg–Shprintzen syndrome (KIAA1279), and Down syndrome.
Epidemiology
Hirschsprung disease occurs in 1 in 5000 live births and has an overall 4:1 male predominance.[8] Conversely, a female predominance in total or extensive colonic disease has been reported in the literature.[12] Hirschsprung disease is a congenital disorder that presents mainly in the neonatal period. The diagnosis is made in 65% of cases before 1 month of age and in 95% before 1 year of age.[13] This disease is rarely diagnosed in adulthood, although the oldest age at presentation reported in the literature is 91 years.
Pathophysiology
Enteric neurons originate from neural crest cells that migrate craniocaudally along the developing gastrointestinal tract, accompanying vagal neural pathways. Colonization of the proximal colon is typically completed by approximately 8 weeks of gestation, with migration reaching the rectum by around 12 weeks. Disturbed rostrocaudal migration of the neural crest cells along a variable length of the intestine is responsible for Hirschsprung disease. Ganglion cells first migrate to the myenteric plexus and then to the submucosal plexus.[14] In Hirschsprung disease, ganglion cells are absent in both the myenteric (Auerbach) and submucosal (Meissner) plexuses of the affected intestinal segment. Animal models have also highlighted an arrest or delay in neural crest cell migration as a factor in the pathogenesis of Hirschsprung disease.[15]
Hirschsprung disease affects the rectosigmoid colon in approximately 80% of patients, representing short-segment disease. The aganglionosis extends proximally to the sigmoid colon in 15% to 20% of patients. This involvement represents long-segment disease. The entire colon is affected in only 5% of cases. This involvement is known as total colonic aganglionosis.
Histopathology
The diagnosis of Hirschsprung disease is based on a combination of clinical features, radiographic findings, and histopathologic evaluation of the biopsied sample. Histopathologic examination of the rectal biopsy confirms the diagnosis by demonstrating the association between the absence of ganglion cells in the submucosal and myenteric plexuses and nerve fiber hypertrophy in the aganglionic segment. Ganglion cells are polygonal cells with abundant fibrillar eosinophilic cytoplasm, an eccentric nucleus, and a large nucleolus.[16] To reduce the rate of inconclusive results due to inadequate biopsies, the International Gastroenterology Committee in 2009 defined criteria for performing preoperative biopsies to ensure reliable interpretation.[17]
At least 2 biopsies are required, with a minimum diameter of 3 mm. The biopsy should have as much submucosa as mucosa. The specimen should be well oriented and aligned with the correct axis to avoid tissue loss across different cutting levels. Preoperative biopsies should be performed at least 2 cm above the dentate line. The distal rectum is physiologically devoid of ganglion cells and exhibits hyperplasia of nerve fibers. Definitive diagnosis or exclusion of Hirschsprung disease on rectal suction biopsy depends on strict histopathologic adequacy and interpretation.[18]
- Clear identification of ganglion cells: A definitive ganglion cell must be recognized based on characteristic morphology, including abundant cytoplasm, an eccentrically placed nucleus, a prominent nucleolus, and perinuclear clearing. Immunohistochemical markers such as Hu antigen C, Hu antigen D, and Phaired-like Homeobox 2B (PHOX2B) may aid in confirmation.
- Proper sampling site: The biopsy should be entirely lined by colonic mucosa to ensure appropriate rectal location.
- Sufficient submucosa: Adequate submucosal tissue must be present in most sections, with a substantial proportion of the specimen composed of submucosa to allow reliable assessment.
- Submucosal nerve hypertrophy: Aganglionic segments characteristically demonstrate increased density and caliber of submucosal nerve fibers, often appearing enlarged compared with the normal distal rectum.
Conventional histopathology with hematoxylin and eosin stain is commonly used in the diagnosis of Hirschsprung disease.[17][19] Acetylcholinesterase (AChE) staining is an ancillary method for identifying increased activity of parasympathetic nerve fibers in the lamina propria and the muscularis mucosae, thereby supporting the diagnosis, especially in difficult cases. Normally, an innervated intestine does not stain with AChE.[20][21] However, AChE staining is laborious and time-consuming, and it requires experienced technicians and pathologists.[21][22] Immunohistochemistry with calretinin antibody has emerged as a useful technique for confirming or ruling out the diagnosis of Hirschsprung disease.[23][24] Calretinin is a vitamin D-dependent protein that binds and buffers calcium in nerve fibers. Loss of calretinin expression leads to intracytoplasmic calcium accumulation, resulting in hyperexcitability and cell degeneration.[15] Calretinin is expressed physiologically in several cell types in human tissues, particularly in the central and peripheral nervous systems.[25]
In 2004, Barschak et al associated the loss of calretinin expression with the absence of ganglion cells, a characteristic of Hirschsprung disease.[12] Immunolabeling with an anticalretinin antibody results in positive findings in ganglion cells and in interstitial nerve fibers in the lamina propria, muscularis mucosae, and submucosa.[26] In the latest recommendations published in 2009, the International Gastroenterology Committee introduced the loss of calretinin expression as one of the diagnostic criteria for Hirschsprung disease.[17]
Frozen section examination is essential in the treatment of Hirschsprung disease and can be performed in 2 circumstances: to establish the diagnosis and to identify the ganglionic zone during endoanal resection.[27][28] Diagnosis during intraoperative evaluation is based on biopsies of only the muscularis of the colonic wall. Circumferential biopsies are increasingly recommended to make a definitive diagnosis.[29]
Choline transporter immunohistochemistry has emerged as a potential alternative to acetylcholinesterase staining.[30] Similar to AChE, the choline transporter highlights the aberrant cholinergic nerve fibers present in aganglionic rectal mucosa. On immunostaining, aganglionic segments demonstrate increased density and caliber of mucosal nerve fibers. This finding is particularly valuable for evaluating biopsies from patients with very short-segment Hirschsprung disease, where diagnostic interpretation may otherwise be challenging.[18]
History and Physical
Several points in the history and physical examination that suggest Hirschsprung disease as a differential diagnosis of neonatal bowel obstruction include (1) abnormal maternal amniotic fluid index, including polyhydramnios; (2) vomiting, specifically bilious emesis; (3) obstipation, which might present with failure to pass meconium in the first 48 hours of life; (4) abdominal distention; and (5) feeding intolerance or failure to thrive. A history of colonic obstruction, which may occur from the early neonatal period to adulthood, along with failure to pass meconium within the first 48 hours of life, which occurs in up to 90% of affected patients, is highly compatible with Hirschsprung disease. However, a history of delayed meconium passage may be present in up to 40% of healthy individuals.[31] Bowel perforation of the cecum, ascending colon, or appendix may be the presenting manifestation in up to 5% of neonates.
Upon rectal examination, patients may have hypertonia of the anal sphincter, and the rectal ampulla is empty. Further clues to a diagnosis of Hirschsprung disease include clinical features of Hirschsprung-associated enterocolitis (HAEC), multiple episodes of overflow constipation, explosive diarrhea (often bloody), vomiting, fever, lethargy, and a soft, distended abdomen. Rare associations with genetic abnormalities, including trisomy 21 and neurocristopathies such as Waardenburg syndrome, have also been reported.[32]
Evaluation
The gold standard for diagnosing Hirschsprung disease is rectal biopsy, with a sensitivity of 93% and a specificity of 98%. Establishing the diagnosis of Hirschsprung disease is generally based on pathologic documentation compatible with the absence of ganglion cells. However, several well-defined obstacles have been identified in obtaining a definitive pathologic diagnosis. These include limited pathologist expertise due to infrequent exposure to samples, inadequate or unacceptable sample quality, disorientation of the submitted tissue in the pathology laboratory, and the presence of normal, immature ganglion cells in newborns.[23]
A key component of the evaluation process in Hirschsprung disease is the assessment of the obtained rectal tissue using specific staining methods, including AChE. However, the crucial role of routine staining methods, including hematoxylin and eosin, should not be underestimated. Fresh-frozen tissue sections might be assessed using AChE staining.[33] Anorectal manometry and contrast enema are other investigations used to aid in diagnosing Hirschsprung disease. Anorectal manometry is based on the principle that the rectoanal inhibitory reflex is uniformly present in all healthy newborns, and the absence of the rectoanal inhibitory reflex on manometry is diagnostic of Hirschsprung disease.[34] In an enema with contrast, features of Hirschsprung disease include a transition zone, a reversal of the rectosigmoid ratio, mucosal irregularity, abnormal contraction, and persistent contrast retention longer than 24 hours (see Image. Hirschsprung Disease).[35]
Treatment / Management
The diagnosis of Hirschsprung disease almost exclusively requires surgical intervention. Pediatric healthcare professionals should possess a comprehensive understanding of the most common surgical procedures to assist in bridging the referral phase between the surgeon and the patient's family. Rectal irrigation before the surgical procedure and in the treatment of Hirschsprung-associated enterocolitis (HAEC) is highly recommended. Rectal irrigation may have 2 crucial advantages, including decompression of the colon and prevention of the most devastating complication, enterocolitis. Surgical planning is significantly affected by the presence of comorbidities, whereas short-segment Hirschsprung disease without any comorbidities can be treated with a single-stage pull-through procedure.[36][37][38][39] In contrast, in the presence of HAEC or a remarkably dilated colon, a staged reconstruction, starting with a temporary decompressive colostomy, should be preferred.[40] (B2)
The recommended timing for a definitive pull-through procedure ranges from 4 to 6 months after colostomy placement. A variety of pull-through procedures have been described. The traditional Swenson technique involves proctectomy, pulling the healthy ganglionated colon through, and anastomosing it to the anus. Novel surgical procedures (eg, Duhamel and Soave procedures) preserve the intricate innervation of the rectum and bladder.[41] In short-segment aganglionosis, the pull-through procedure may be performed using a transanal approach. An abdominal component, either laparoscopic or open, is frequently incorporated to accurately determine the proximal extent of aganglionosis before commencing the pull-through and to assist with colonic mobilization when required. The early postoperative period following the Soave procedure is critical. Regular sessions of mechanical anastomotic dilation, which may be performed at home, are highly recommended. All of these procedures have high success rates, and morbidity is minimal.[42] An alternative approach is to perform a single-stage transanal Soave procedure early in the neonatal period, which might obviate the need for an abdominal incision and colostomy.[43] However, the complication rates are quite similar to those of the more invasive procedures.[37][41][42][44](B2)
Differential Diagnosis
Hirschsprung disease has been classified as an intestinal dysganglionosis and should be differentiated from intestinal neuronal dysplasia (IND) and isolated hypoganglionosis. The diagnostic criteria for IND have evolved since 1992, and the following diagnostic indices by Borchard et al are widely accepted: (1) abnormally hyperplastic submucosal plexus and (2) enhanced perivascular AChE activity in the submucosal layer. Several additional criteria may be used to confirm IND, including heterotopic ganglion cells and abnormally enhanced acetylcholinesterase activity in the lamina propria and the circular muscular layer.[45]
Moreover, Hirschsprung disease should be distinguished from the wide spectrum of Hirschsprung disease variants that clinically mimic its signs and symptoms. This spectrum includes not only IND but also other disorders with ganglion cells in rectal biopsies, including intestinal ganglioneuromatosis, isolated hypoganglionosis, immature ganglia, absence of the argyrophil plexus, internal anal sphincter achalasia, and megacystis-microcolon intestinal hypoperistalsis syndrome. Internal anal sphincter achalasia may be distinguished from the others, with the only pathologic finding on anorectal manometry suggesting the absence of the rectosphincteric reflex. Using specific staining of the rectal biopsy specimen may be essential to differentiate the remaining pathologies within the spectrum. Intestinal ganglioneuromatosis, which is mainly confined to the colorectal area and may spare the small bowel, can be differentiated based on the extraordinary proliferation of both the submucosal and myenteric plexuses, comprising thick nerve trunks and accompanied by scattered mature neurons, giant ganglia, and enhanced AChE activity.[46][47][48]
Isolated hypoganglionosis, one of the rarest differential diagnoses in this era, may be suggested by immunohistochemical staining.[49] Immature ganglia are a physiologic, age-dependent spectrum that presents with chronic constipation. The normal aging course may reverse the small ganglion cells, which are more evident on nicotinamide adenine dinucleotide phosphate-diaphorase and neural cell adhesion molecule staining than on routine AChE histochemistry.[49][50] The absence of the argyrophilic plexus, a rare differential diagnosis of Hirschsprung disease, may reflect disturbed differentiation of argyrophilic cells.[51]
Megacystis-microcolon intestinal hypoperistalsis syndrome is categorized as one of the least common disorders in this spectrum. This syndrome may cause the most severe clinical presentations of neonatal functional obstruction syndromes. Several hypotheses have been proposed to identify the exact mechanistic pathways responsible. Although the definitive description has not yet been well established, mutations in the nicotinic acetylcholine receptor and reduced expression of the α3, β2, and β4 subunits of the nicotinic acetylcholine receptor in small bowel tissue might share similar clinical manifestations.[52][53][54]
Systemic disorders such as hypothyroidism, primary myopathies, and neuropathies can impair gastrointestinal tract motility and mimic functional obstruction. Structural causes, including colonic or distal small intestinal atresia and stenosis, may present in the neonatal period with symptoms resembling Hirschsprung disease. Neonatal obstruction secondary to inspissated meconium is typically categorized as meconium ileus, meconium plug syndrome, or meconium disease. Although diagnostic criteria overlap and terminology is inconsistently applied, these entities may initially be indistinguishable from Hirschsprung disease. In older infants and children, functional constipation remains a common consideration. Careful assessment of symptom onset is critical, particularly the timing of first meconium passage, because delayed passage in the neonatal period strongly raises suspicion for Hirschsprung disease.
Prognosis
The quality of life in patients with Hirschsprung disease depends on the degree of fecal continence. Literature remains limited on the quality of life in long-standing Hirschsprung disease. In one unique, comprehensive, nationwide French survey, approximately 3000 adult patients with Hirschsprung disease were evaluated using a modified questionnaire.[55] The original questionnaire was described by a Dutch team in 2001 and addressed not only Hirschsprung disease but also the wide spectrum of anorectal malformations.[56]
Results from a systematic review and meta-analysis showed pooled prevalences of 20% for fecal incontinence, 14% for constipation, and 7% for bladder dysfunction. Most patients had good bowel function scores but lower quality of life in the lower gastrointestinal tract compared with the general population. This systematic review and meta-analysis included 12 studies and 625 patients older than 10 years and evaluated long-term functional outcomes and quality of life after surgical procedures for Hirschsprung disease.[57]
Complications
One of the most common and devastating Hirschsprung disease-related complications is Hirschsprung-associated enterocolitis (HAEC), which is defined as an inflammatory disorder of the bowel. To obtain the diagnosis of HAEC, which is the most challenging, a combination of clinical signs and symptoms should be considered; this will result in an inevitable over- and under-treatment of patients. The underlying cause of HAEC remains ill-defined. Several hypotheses, including dysbiosis of the intestinal microbiome,[58][59] impaired mucosal barrier function,[32][60][61] altered innate immune responses,[62] and bacterial translocation, have been proposed.[63][64]
The severity of clinical manifestations in HAEC may vary, and several scoring systems have been developed to classify gastrointestinal tract and systemic presentations, including a Delphi method to reach consensus among a panel of pediatric gastroenterologists and surgeons, and a clinical grading system based on a prospective trial. The former uses 16 items, whereas the latter is based on 3 main components: diarrhea, abdominal distention, and systemic manifestations to define the grading of HAEC.[65] Treatment of each patient with HAEC is based on the corresponding clinical grade.
In grade 1, or possible HAEC, outpatient treatment with oral metronidazole, accompanied by fluid and electrolyte replacement, may be considered. More severe cases, including definite and severe HAEC, should be admitted to the hospital and treated with intravenous fluid resuscitation and broad-spectrum antibiotics. Rectal irrigation to remove retained stool and reduce the bacterial load may be considered in patients with abdominal distention, regardless of HAEC grade. Surgical intervention with a proximal colostomy may be considered in children with severe HAEC who do not respond to primary medical treatment with bowel rest, intravenous fluid resuscitation, rectal irrigations, and broad-spectrum antibiotics.[32] Several other less frequent postoperative complications of Hirschsprung disease may occur, including an anastomotic leak in up to 1.5% of patients, anastomotic stricture, wound infections, bleeding, and perianal excoriations.[40][65][66] Several abnormal variations in bowel habits, including constipation and fecal incontinence, may occur following a variety of well-defined surgical procedures for Hirschsprung disease. The former may be attributed to high anal resting pressure and weak rectal peristalsis, whereas the latter is the result of poor surgical technique.[66]
Postoperative and Rehabilitation Care
Children with Hirschsprung disease remain at lifelong risk of incontinence, enterocolitis, and obstructive symptoms, regardless of surgical technique. Structured interdisciplinary follow-up, more frequent in infancy and annually thereafter, is essential to monitor bowel function, growth, nutrition, and psychosocial development. Care should address broader outcomes, including self-efficacy, coping, and sexual health.[67]
Deterrence and Patient Education
Although the most worrisome consequences of Hirschsprung disease may not persist into adulthood, a significant proportion of affected patients who have undergone definitive surgical procedures during infancy may experience bowel habit changes during school age. These complications may cause significant psychological morbidity, considerable changes in quality of life, and parental challenges, including work leave. Therefore, using an interprofessional team approach to ultimately toilet train the child is crucial and requires several essential steps, including training in optimal defecation skills and toilet behavior.[68]
Parents should be aware of any suspicious symptoms suggestive of Hirschsprung disease, including a delay in meconium passage of more than 48 hours during the neonatal period. Moreover, nonspecific symptoms, including constipation, fecal incontinence and pseudoincontinence, abdominal distention, reflux, nausea, vomiting, and diarrhea, should also raise suspicion.[69] Parents should be counseled and made aware of the need for staged or single-stage surgical procedures and the complications associated with each approach. A subset of patients may develop ongoing constipation requiring dietary modification, including increased fiber intake, and pharmacologic support with laxatives.
Enhancing Healthcare Team Outcomes
An interprofessional team approach to treatment based on a biopsychosocial view can significantly reduce postoperative chronic bowel problems in most children after surgical treatment for Hirschsprung disease. The central aim is to teach self-control: the more the child can evacuate adequate amounts of feces regularly and voluntarily, the less medication the child will need and the more independent the child will be from the clinician and parents. Solving defecation issues at an early age prevents invasive procedures from the start of treatment, which is important given the potential harms of procedures in a sensitive region, especially during the child's early development.
Depending on the length of the intestine that needs to be removed, children with Hirschsprung disease may have varying levels of long-term care needs. Temporary or intermittent problems may include frequent and loose stools, difficulty sensing the need to have a bowel movement, and problems with peristalsis because the anal opening is tight. From a nutritional perspective, removing a large portion of the intestine may prevent a child from receiving adequate nutrients and fluids. Children may experience improper digestion, slow growth, and infection.
Nutritionists help meet nutritional goals and offer suggestions to support nutritional requirements, ensuring the child grows and develops normally. While the pediatric surgeon is almost always involved in the treatment of patients with Hirschsprung disease, an interprofessional team of subspecialists should also be involved, including a pediatric gastroenterologist, pediatric radiologist, and a specialist in bowel care protocols. Experienced nurses are also crucial members of the group because they monitor the patient's vital signs and warning signs of potentially devastating complications, including Hirschsprung-associated enterocolitis, and assist in educating the patient and family. Nurses also provide crucial education on bowel, skin, and stoma care. In the postoperative period, the pharmacist ensures that the patient is prescribed the correct analgesics, antiemetics, and appropriate antibiotics.[69] Psychosocial support is crucial for addressing the trauma of diagnosis and long-term care needs, including school accommodations.
Media
(Click Image to Enlarge)
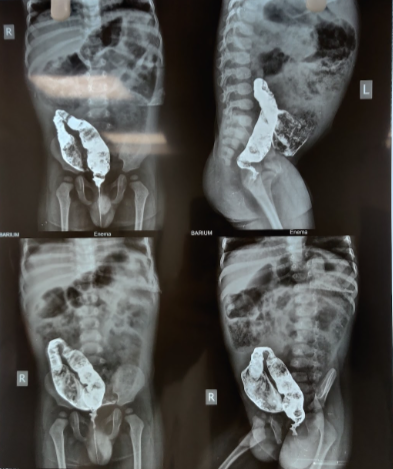
Hirschsprung Disease. Rectal contrast enema showing collapsed rectum, with dilated sigmoid colon. The reversal of the recto-sigmoid ratio (RS ratio) is also evident.
Contributed by K Kulkarni, PA
References
Martucciello G. Hirschsprung's disease, one of the most difficult diagnoses in pediatric surgery: a review of the problems from clinical practice to the bench. European journal of pediatric surgery : official journal of Austrian Association of Pediatric Surgery ... [et al] = Zeitschrift fur Kinderchirurgie. 2008 Jun:18(3):140-9. doi: 10.1055/s-2008-1038625. Epub [PubMed PMID: 18493886]
Drabent P, Bonnard A, Guimiot F, Peuchmaur M, Berrebi D. PHOX2B Immunostaining: A Simple and Helpful Tool for the Recognition of Ganglionic Cells and Diagnosis of Hirschsprung Disease. The American journal of surgical pathology. 2020 Oct:44(10):1389-1397. doi: 10.1097/PAS.0000000000001528. Epub [PubMed PMID: 32604166]
Meza-Valencia BE, de Lorimier AJ, Person DA. Hirschsprung disease in the U.S. associated Pacific Islands: more common than expected. Hawaii medical journal. 2005 Apr:64(4):96-8, 100-1 [PubMed PMID: 15921246]
Coi A, Santoro M, Pierini A, Rankin J, Glinianaia SV, Tan J, Reid AK, Garne E, Loane M, Given J, Ballardini E, Cavero-Carbonell C, de Walle HEK, Gatt M, García-Villodre L, Gissler M, Jordan S, Kiuru-Kuhlefelt S, Kjaer Urhoj S, Klungsøyr K, Lelong N, Lutke LR, Neville AJ, Rahshenas M, Scanlon I, Wellesley D, Morris JK. Survival of children with rare structural congenital anomalies: a multi-registry cohort study. Orphanet journal of rare diseases. 2022 Mar 29:17(1):142. doi: 10.1186/s13023-022-02292-y. Epub 2022 Mar 29 [PubMed PMID: 35351164]
Samujh R. Hirschsprung's Disease: Perspectives upon Late Presentation in India and Developing Nations. Journal of Indian Association of Pediatric Surgeons. 2022 May-Jun:27(3):275-278. doi: 10.4103/jiaps.jiaps_6_22. Epub 2022 May 12 [PubMed PMID: 35733593]
Level 3 (low-level) evidenceBhatnagar SN. Hirschsprung's Disease in Newborns. Journal of neonatal surgery. 2013 Oct-Dec:2(4):51 [PubMed PMID: 26023471]
Burns AJ, Goldstein AM. Causes and consequences: development and pathophysiology of Hirschsprung disease. World journal of pediatric surgery. 2024:7(4):e000903. doi: 10.1136/wjps-2024-000903. Epub 2024 Nov 25 [PubMed PMID: 39600627]
Butler Tjaden NE, Trainor PA. The developmental etiology and pathogenesis of Hirschsprung disease. Translational research : the journal of laboratory and clinical medicine. 2013 Jul:162(1):1-15. doi: 10.1016/j.trsl.2013.03.001. Epub 2013 Mar 22 [PubMed PMID: 23528997]
Level 3 (low-level) evidenceKarim A, Tang CS, Tam PK. The Emerging Genetic Landscape of Hirschsprung Disease and Its Potential Clinical Applications. Frontiers in pediatrics. 2021:9():638093. doi: 10.3389/fped.2021.638093. Epub 2021 Aug 5 [PubMed PMID: 34422713]
Löf Granström A, Wester T. Mortality in Swedish patients with Hirschsprung disease. Pediatric surgery international. 2017 Nov:33(11):1177-1181. doi: 10.1007/s00383-017-4150-z. Epub 2017 Sep 7 [PubMed PMID: 28884210]
Núñez-Ramos R, Fernández RM, González-Velasco M, Ruiz-Contreras J, Galán-Gómez E, Núñez-Núñez R, Borrego S. A Scoring System to Predict the Severity of Hirschsprung Disease at Diagnosis and Its Correlation With Molecular Genetics. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2017 Jan-Feb:20(1):28-37. doi: 10.1177/1093526616683883. Epub [PubMed PMID: 28276298]
Barshack I, Fridman E, Goldberg I, Chowers Y, Kopolovic J. The loss of calretinin expression indicates aganglionosis in Hirschsprung's disease. Journal of clinical pathology. 2004 Jul:57(7):712-6 [PubMed PMID: 15220363]
Harrison MW, Deitz DM, Campbell JR, Campbell TJ. Diagnosis and management of Hirschsprung's disease. A 25 year perspective. American journal of surgery. 1986 Jul:152(1):49-56 [PubMed PMID: 3728817]
Level 3 (low-level) evidenceDasgupta R, Langer JC. Hirschsprung disease. Current problems in surgery. 2004 Dec:41(12):942-88 [PubMed PMID: 15614238]
Webster W. Embryogenesis of the enteric ganglia in normal mice and in mice that develop congenital aganglionic megacolon. Journal of embryology and experimental morphology. 1973 Dec:30(3):573-85 [PubMed PMID: 4772386]
Level 3 (low-level) evidenceGonzalo DH, Plesec T. Hirschsprung disease and use of calretinin in inadequate rectal suction biopsies. Archives of pathology & laboratory medicine. 2013 Aug:137(8):1099-102. doi: 10.5858/arpa.2012-0220-OA. Epub [PubMed PMID: 23899067]
Level 2 (mid-level) evidenceKnowles CH, De Giorgio R, Kapur RP, Bruder E, Farrugia G, Geboes K, Gershon MD, Hutson J, Lindberg G, Martin JE, Meier-Ruge WA, Milla PJ, Smith VV, Vandervinden JM, Veress B, Wedel T. Gastrointestinal neuromuscular pathology: guidelines for histological techniques and reporting on behalf of the Gastro 2009 International Working Group. Acta neuropathologica. 2009 Aug:118(2):271-301. doi: 10.1007/s00401-009-0527-y. Epub 2009 Apr 10 [PubMed PMID: 19360428]
Ambartsumyan L, Smith C, Kapur RP. Diagnosis of Hirschsprung Disease. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2020 Jan-Feb:23(1):8-22. doi: 10.1177/1093526619892351. Epub 2019 Dec 2 [PubMed PMID: 31791203]
Qualman SJ, Jaffe R, Bove KE, Monforte-Muñoz H. Diagnosis of hirschsprung disease using the rectal biopsy: multi-institutional survey. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 1999 Nov-Dec:2(6):588-96 [PubMed PMID: 10508885]
Level 3 (low-level) evidenceSantos MM, Tannuri U, Coelho MC. Study of acetylcholinesterase activity in rectal suction biopsy for diagnosis of intestinal dysganglionoses: 17-year experience of a single center. Pediatric surgery international. 2008 Jun:24(6):715-9. doi: 10.1007/s00383-008-2141-9. Epub 2008 Apr 12 [PubMed PMID: 18408941]
Moore SW, Johnson G. Acetylcholinesterase in Hirschsprung's disease. Pediatric surgery international. 2005 Apr:21(4):255-63 [PubMed PMID: 15759143]
Rakhshani N, Araste M, Imanzade F, Panahi M, Safarnezhad Tameshkel F, Sohrabi MR, Karbalaie Niya MH, Zamani F. Hirschsprung Disease Diagnosis: Calretinin Marker Role in Determining the Presence or Absence of Ganglion Cells. Iranian journal of pathology. 2016 Fall:11(4):409-415 [PubMed PMID: 28855933]
Cinel L, Ceyran B, Güçlüer B. Calretinin immunohistochemistry for the diagnosis of Hirschprung disease in rectal biopsies. Pathology, research and practice. 2015 Jan:211(1):50-4. doi: 10.1016/j.prp.2014.08.012. Epub 2014 Nov 4 [PubMed PMID: 25442014]
Jiang M, Li K, Li S, Yang L, Yang D, Zhang X, Fang M, Cao G, Wang Y, Chen W, Tang S. Calretinin, S100 and protein gene product 9.5 immunostaining of rectal suction biopsies in the diagnosis of Hirschsprung' disease. American journal of translational research. 2016:8(7):3159-68 [PubMed PMID: 27508037]
Guinard-Samuel V, Bonnard A, De Lagausie P, Philippe-Chomette P, Alberti C, El Ghoneimi A, Peuchmaur M, Berrebi-Binczak D. Calretinin immunohistochemistry: a simple and efficient tool to diagnose Hirschsprung disease. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2009 Oct:22(10):1379-84. doi: 10.1038/modpathol.2009.110. Epub 2009 Jul 31 [PubMed PMID: 19648883]
Level 2 (mid-level) evidencede Haro Jorge I, Palazón Bellver P, Julia Masip V, Saura García L, Ribalta Farres T, Cuadras Pallejà D, Tarrado Castellarnau X. Effectiveness of calretinin and role of age in the diagnosis of Hirschsprung disease. Pediatric surgery international. 2016 Aug:32(8):723-7. doi: 10.1007/s00383-016-3912-3. Epub 2016 Jul 1 [PubMed PMID: 27369965]
Ghose SI, Squire BR, Stringer MD, Batcup G, Crabbe DC. Hirschsprung's disease: problems with transition-zone pull-through. Journal of pediatric surgery. 2000 Dec:35(12):1805-9 [PubMed PMID: 11101741]
Level 2 (mid-level) evidenceMaia DM. The reliability of frozen-section diagnosis in the pathologic evaluation of Hirschsprung's disease. The American journal of surgical pathology. 2000 Dec:24(12):1675-7 [PubMed PMID: 11117790]
Kapur RP, Kennedy AJ. Transitional zone pull through: surgical pathology considerations. Seminars in pediatric surgery. 2012 Nov:21(4):291-301. doi: 10.1053/j.sempedsurg.2012.07.003. Epub [PubMed PMID: 22985834]
Kapur RP, Raess PW, Hwang S, Winter C. Choline Transporter Immunohistochemistry: An Effective Substitute for Acetylcholinesterase Histochemistry to Diagnose Hirschsprung Disease With Formalin-fixed Paraffin-embedded Rectal Biopsies. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2017 Jul-Aug:20(4):308-320. doi: 10.1177/1093526617697060. Epub 2017 Mar 23 [PubMed PMID: 28649946]
de Lorijn F, Boeckxstaens GE, Benninga MA. Symptomatology, pathophysiology, diagnostic work-up, and treatment of Hirschsprung disease in infancy and childhood. Current gastroenterology reports. 2007 Jun:9(3):245-53 [PubMed PMID: 17511924]
Gosain A, Frykman PK, Cowles RA, Horton J, Levitt M, Rothstein DH, Langer JC, Goldstein AM, American Pediatric Surgical Association Hirschsprung Disease Interest Group. Guidelines for the diagnosis and management of Hirschsprung-associated enterocolitis. Pediatric surgery international. 2017 May:33(5):517-521. doi: 10.1007/s00383-017-4065-8. Epub 2017 Feb 2 [PubMed PMID: 28154902]
Rabah R. Total colonic aganglionosis: case report, practical diagnostic approach and pitfalls. Archives of pathology & laboratory medicine. 2010 Oct:134(10):1467-73 [PubMed PMID: 20923302]
Level 3 (low-level) evidenceTang YF, Chen JG, An HJ, Jin P, Yang L, Dai ZF, Huang LM, Yu JW, Yang XY, Fan RY, Li SJ, Han Y, Wang JH, Gyawali CP, Sheng JQ. High-resolution anorectal manometry in newborns: normative values and diagnostic utility in Hirschsprung disease. Neurogastroenterology and motility. 2014 Nov:26(11):1565-72. doi: 10.1111/nmo.12423. Epub 2014 Sep 27 [PubMed PMID: 25263969]
Peyvasteh M, Askarpour S, Ostadian N, Moghimi MR, Javaherizadeh H. DIAGNOSTIC ACCURACY OF BARIUM ENEMA FINDINGS IN HIRSCHSPRUNG'S DISEASE. Arquivos brasileiros de cirurgia digestiva : ABCD = Brazilian archives of digestive surgery. 2016 Jul-Sep:29(3):155-158. doi: 10.1590/0102-6720201600030007. Epub [PubMed PMID: 27759777]
Coran AG, Teitelbaum DH. Recent advances in the management of Hirschsprung's disease. American journal of surgery. 2000 Nov:180(5):382-7 [PubMed PMID: 11137692]
Level 3 (low-level) evidenceWeidner BC, Waldhausen JH. Swenson revisited: a one-stage, transanal pull-through procedure for Hirschsprung's disease. Journal of pediatric surgery. 2003 Aug:38(8):1208-11 [PubMed PMID: 12891494]
Rintala RJ. Transanal coloanal pull-through with a short muscular cuff for classic Hirschsprung's disease. European journal of pediatric surgery : official journal of Austrian Association of Pediatric Surgery ... [et al] = Zeitschrift fur Kinderchirurgie. 2003 Jun:13(3):181-6 [PubMed PMID: 12939703]
Level 2 (mid-level) evidenceHadidi A. Transanal endorectal pull-through for Hirschsprung's disease: a comparison with the open technique. European journal of pediatric surgery : official journal of Austrian Association of Pediatric Surgery ... [et al] = Zeitschrift fur Kinderchirurgie. 2003 Jun:13(3):176-80 [PubMed PMID: 12939702]
Langer JC, Durrant AC, de la Torre L, Teitelbaum DH, Minkes RK, Caty MG, Wildhaber BE, Ortega SJ, Hirose S, Albanese CT. One-stage transanal Soave pullthrough for Hirschsprung disease: a multicenter experience with 141 children. Annals of surgery. 2003 Oct:238(4):569-83; discussion 583-5 [PubMed PMID: 14530728]
Level 2 (mid-level) evidenceHadidi A. Transanal endorectal pull-through for Hirschsprung's disease: experience with 68 patients. Journal of pediatric surgery. 2003 Sep:38(9):1337-40 [PubMed PMID: 14523816]
Saleh W, Rasheed K, Mohaidly MA, Kfoury H, Tariq M, Rawaf AA. Management of Hirschsprung's disease: a comparison of Soave's and Duhamel's pull-through methods. Pediatric surgery international. 2004 Aug:20(8):590-3 [PubMed PMID: 15309470]
Level 2 (mid-level) evidenceEkema G, Falchetti D, Torri F, Merulla VE, Manciana A, Caccia G. Further evidence on totally transanal one-stage pull-through procedure for Hirschsprung's disease. Journal of pediatric surgery. 2003 Oct:38(10):1434-9 [PubMed PMID: 14577064]
Tomita R, Ikeda T, Fujisaki S, Tanjoh K, Munakata K. Hirschsprung's disease and its allied disorders in adults' histological and clinical studies. Hepato-gastroenterology. 2003 Jul-Aug:50(52):1050-3 [PubMed PMID: 12845979]
Pini-Prato A, Martucciello G, Jasonni V. Rectal suction biopsy in the diagnosis of intestinal dysganglionoses: 5-year experience with Solo-RBT in 389 patients. Journal of pediatric surgery. 2006 Jun:41(6):1043-8 [PubMed PMID: 16769331]
Plenat F, Vignaud JM, Floquet J, Leroux P, Briquel N, Morali A, Vidailhet M. [Intestinal ganglioneuromatosis: histochemical, histoenzymological and ultrastructural study of a case]. Annales de pathologie. 1984 Apr-May:4(2):131-6 [PubMed PMID: 6732905]
Level 3 (low-level) evidenceSmith VV, Eng C, Milla PJ. Intestinal ganglioneuromatosis and multiple endocrine neoplasia type 2B: implications for treatment. Gut. 1999 Jul:45(1):143-6 [PubMed PMID: 10369718]
Level 3 (low-level) evidenceTorre M, Martucciello G, Ceccherini I, Lerone M, Aicardi M, Gambini C, Jasonni V. Diagnostic and therapeutic approach to multiple endocrine neoplasia type 2B in pediatric patients. Pediatric surgery international. 2002 Sep:18(5-6):378-83 [PubMed PMID: 12415360]
Level 3 (low-level) evidencePuri P, Fujimoto T. Diagnosis of allied functional bowel disorders using monoclonal antibodies and electronmicroscopy. Journal of pediatric surgery. 1988 Jun:23(6):546-54 [PubMed PMID: 3418474]
Puri P. Variant Hirschsprung's disease. Journal of pediatric surgery. 1997 Feb:32(2):149-57 [PubMed PMID: 9044113]
SINGH I. THE PRENATAL DEVELOPMENT OF ENTEROCHROMAFFIN CELLS IN THE HUMAN GASTRO-INTESTINAL TRACT. Journal of anatomy. 1963 Jul:97(Pt 3):377-87 [PubMed PMID: 14047355]
Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou CN, Patrick J, Role L, De Biasi M, Beaudet AL. Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proceedings of the National Academy of Sciences of the United States of America. 1999 May 11:96(10):5746-51 [PubMed PMID: 10318955]
Level 3 (low-level) evidenceRichardson CE, Morgan JM, Jasani B, Green JT, Rhodes J, Williams GT, Lindstrom J, Wonnacott S, Thomas GA, Smith V. Megacystis-microcolon-intestinal hypoperistalsis syndrome and the absence of the alpha3 nicotinic acetylcholine receptor subunit. Gastroenterology. 2001 Aug:121(2):350-7 [PubMed PMID: 11487544]
Xu W, Orr-Urtreger A, Nigro F, Gelber S, Sutcliffe CB, Armstrong D, Patrick JW, Role LW, Beaudet AL, De Biasi M. Multiorgan autonomic dysfunction in mice lacking the beta2 and the beta4 subunits of neuronal nicotinic acetylcholine receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1999 Nov 1:19(21):9298-305 [PubMed PMID: 10531434]
Level 3 (low-level) evidenceDrissi F, Meurette G, Baayen C, Wyart V, Cretolle C, Guinot A, Podevin G, Lehur PA. Long-term Outcome of Hirschsprung Disease: Impact on Quality of Life and Social Condition at Adult Age. Diseases of the colon and rectum. 2019 Jun:62(6):727-732. doi: 10.1097/DCR.0000000000001363. Epub [PubMed PMID: 30807458]
Level 2 (mid-level) evidenceHartman EE, Oort FJ, Sprangers MA, Hanneman MJ, van Heurn LW, de Langen ZJ, Madern GC, Rieu PN, van der Zee DC, Looyaard N, van Silfhout-Bezemer M, Aronson DC. Factors affecting quality of life of children and adolescents with anorectal malformations or Hirschsprung disease. Journal of pediatric gastroenterology and nutrition. 2008 Oct:47(4):463-71. doi: 10.1097/MPG.0b013e31815ce545. Epub [PubMed PMID: 18852639]
Level 2 (mid-level) evidenceDai Y, Deng Y, Lin Y, Ouyang R, Li L. Long-term outcomes and quality of life of patients with Hirschsprung disease: a systematic review and meta-analysis. BMC gastroenterology. 2020 Mar 12:20(1):67. doi: 10.1186/s12876-020-01208-z. Epub 2020 Mar 12 [PubMed PMID: 32164539]
Level 1 (high-level) evidenceWard NL, Pieretti A, Dowd SE, Cox SB, Goldstein AM. Intestinal aganglionosis is associated with early and sustained disruption of the colonic microbiome. Neurogastroenterology and motility. 2012 Sep:24(9):874-e400. doi: 10.1111/j.1365-2982.2012.01937.x. Epub 2012 May 24 [PubMed PMID: 22626027]
Level 3 (low-level) evidenceFrykman PK, Nordenskjöld A, Kawaguchi A, Hui TT, Granström AL, Cheng Z, Tang J, Underhill DM, Iliev I, Funari VA, Wester T, HAEC Collaborative Research Group (HCRG). Characterization of Bacterial and Fungal Microbiome in Children with Hirschsprung Disease with and without a History of Enterocolitis: A Multicenter Study. PloS one. 2015:10(4):e0124172. doi: 10.1371/journal.pone.0124172. Epub 2015 Apr 24 [PubMed PMID: 25909773]
Level 2 (mid-level) evidenceThiagarajah JR, Yildiz H, Carlson T, Thomas AR, Steiger C, Pieretti A, Zukerberg LR, Carrier RL, Goldstein AM. Altered goblet cell differentiation and surface mucus properties in Hirschsprung disease. PloS one. 2014:9(6):e99944. doi: 10.1371/journal.pone.0099944. Epub 2014 Jun 19 [PubMed PMID: 24945437]
Level 3 (low-level) evidenceYildiz HM, Carlson TL, Goldstein AM, Carrier RL. Mucus Barriers to Microparticles and Microbes are Altered in Hirschsprung's Disease. Macromolecular bioscience. 2015 May:15(5):712-8. doi: 10.1002/mabi.201400473. Epub 2015 Feb 2 [PubMed PMID: 25644515]
Level 3 (low-level) evidenceFrykman PK, Cheng Z, Wang X, Dhall D. Enterocolitis causes profound lymphoid depletion in endothelin receptor B- and endothelin 3-null mouse models of Hirschsprung-associated enterocolitis. European journal of immunology. 2015 Mar:45(3):807-17. doi: 10.1002/eji.201444737. Epub 2015 Jan 19 [PubMed PMID: 25487064]
Level 3 (low-level) evidencePierre JF, Barlow-Anacker AJ, Erickson CS, Heneghan AF, Leverson GE, Dowd SE, Epstein ML, Kudsk KA, Gosain A. Intestinal dysbiosis and bacterial enteroinvasion in a murine model of Hirschsprung's disease. Journal of pediatric surgery. 2014 Aug:49(8):1242-51. doi: 10.1016/j.jpedsurg.2014.01.060. Epub [PubMed PMID: 25092084]
Level 3 (low-level) evidenceCheng Z, Dhall D, Zhao L, Wang HL, Doherty TM, Bresee C, Frykman PK. Murine model of Hirschsprung-associated enterocolitis. I: phenotypic characterization with development of a histopathologic grading system. Journal of pediatric surgery. 2010 Mar:45(3):475-82. doi: 10.1016/j.jpedsurg.2009.06.009. Epub [PubMed PMID: 20223308]
Level 3 (low-level) evidenceBjørnland K, Pakarinen MP, Stenstrøm P, Stensrud KJ, Neuvonen M, Granström AL, Graneli C, Pripp AH, Arnbjörnsson E, Emblem R, Wester T, Rintala RJ, Nordic Pediatric Surgery Study Consortium. A Nordic multicenter survey of long-term bowel function after transanal endorectal pull-through in 200 patients with rectosigmoid Hirschsprung disease. Journal of pediatric surgery. 2017 Sep:52(9):1458-1464. doi: 10.1016/j.jpedsurg.2017.01.001. Epub 2017 Jan 5 [PubMed PMID: 28094015]
Level 3 (low-level) evidenceKeshtgar AS, Ward HC, Clayden GS, de Sousa NM. Investigations for incontinence and constipation after surgery for Hirschsprung's disease in children. Pediatric surgery international. 2003 Apr:19(1-2):4-8 [PubMed PMID: 12721712]
Kyrklund K, Sloots CEJ, de Blaauw I, Bjørnland K, Rolle U, Cavalieri D, Francalanci P, Fusaro F, Lemli A, Schwarzer N, Fascetti-Leon F, Thapar N, Johansen LS, Berrebi D, Hugot JP, Crétolle C, Brooks AS, Hofstra RM, Wester T, Pakarinen MP. ERNICA guidelines for the management of rectosigmoid Hirschsprung's disease. Orphanet journal of rare diseases. 2020 Jun 25:15(1):164. doi: 10.1186/s13023-020-01362-3. Epub 2020 Jun 25 [PubMed PMID: 32586397]
van Kuyk EM, Brugman-Boezeman AT, Wissink-Essink M, Severijnen RS, Festen C, Bleijenberg G. Defecation problems in children with Hirschsprung's disease: a biopsychosocial approach. Pediatric surgery international. 2000:16(5-6):312-6 [PubMed PMID: 10955552]
Level 2 (mid-level) evidenceAmerican Academy of Family Physicians. Information from your family doctor. Hirschsprung's disease: what you should know. American family physician. 2006 Oct 15:74(8):1327-8 [PubMed PMID: 17087426]