Introduction
Hepatoblastomas are the most common primary malignant liver tumor in pediatric patients, occurring mostly within the first 2 years of life.[1] The histologic types are subdivided into 2 broad categories: epithelial and mixed. Over the last 3 decades, the treatment has advanced, with neoadjuvant chemotherapy now the standard of care for most cases. Neoadjuvant chemotherapy and surgical resection yield an approximately 70% cure rate, a vast improvement over the dismal 30% in the 1970s. Prognosis is based on many factors, including α-fetoprotein levels, age at diagnosis, completeness of resection, and clinical stage.[2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Most tumors are sporadic, but one-third of cases may be associated with Beckwith-Weidemann syndrome, familial adenomatous polyposis, Edward syndrome (trisomy 18), nephroblastoma, and Down syndrome.[3] Low birth weight infants are at higher risk of developing a hepatoblastoma, and evidence has shown an association with preeclampsia and parental tobacco smoking before and during pregnancy.[3][4] Other factors thought to play a role in pathogenesis include oxygen therapy, certain medications (furosemide), radiation, plasticizers, and total parenteral nutrition.[2]
The most common genetic mutation involves the Wnt signaling pathway, which results in the accumulation of beta-catenin; these mutations are present at a higher proportion in sporadic cases.[5] By immunohistochemistry, β-catenin usually shows a membranous staining pattern in the more differentiated fetal types and a nuclear staining pattern in the less differentiated histologic types.[6] Results from studies showed activation of the human telomerase reverse transcriptase gene (TERT) and MYC signaling in aggressive cases.[2]
Epidemiology
Hepatoblastoma is a rare tumor comprising approximately 1% of all pediatric tumors.[7] The incidence rate is slowly increasing in North America and Europe, and there is a slight male predominance.[8]
Histopathology
Hepatoblastomas originate from primitive hepatic stem cells that give rise to the liver's epithelial components. Usually, these tumors are divided into 2 broad categories: epithelial type and mixed epithelial and mesenchymal type (MEM-HB). Revision of the original classification system led to the pathology consensus for the pediatric hepatoblastoma classification system, which retained the subdivision of histologic types into 2 broad categories, as described above. The E-HB includes fetal, pleomorphic, embryonal, macrotrabecular, small cell undifferentiated (SCU), cholangioblastic, and mixed epithelial variants. The MEM-HB is subdivided into tumors with teratoid features and those without.
The fetal subtype is further stratified into 4 categories: well-differentiated; crowded or mitotically active; pleomorphic, poorly differentiated; and anaplastic. The well-differentiated variant is characterized by a low-power view demonstrating alternating light and dark areas due to variable cytoplasmic glycogen content. Assessment at higher magnification reveals a uniform population of hepatocytes arranged in trabeculae 2 to 3 cells thick. Extramedullary hematopoiesis is a typical finding, and mitotic rate is low. Description of the other variants is beyond the scope of this article.
The embryonal subtype is the most commonly encountered and consists of basophilic cells with scant cytoplasm and an increased mitotic rate, arranged in nests, trabeculae, acini, pseudorosettes, or sheets. The macrotrabecular subtype is composed of trabeculae more than 10 cells thick. The SCU subtype consists of dyscohesive, uniform round cells arranged in sheets with increased mitotic activity. Some cases of SCU have a loss of INI1, suggesting a possible association with primary rhabdoid tumors of the liver. The cholangioblastic variant has bile ducts, typically located at the periphery of epithelial sheets.
The MEM-HB comprises 20% to 30% of tumors and contains a variable combination of epithelial and mesenchymal components. Most commonly, the epithelial component is fetal or embryonal, and the mesenchymal component is osteoid. Stromal derivatives include spindle cells, osteoid, skeletal muscle, and cartilage. Teratoid features include primitive endoderm, neural derivatives, melanin, squamous and glandular elements.[2][9]
History and Physical
Hepatoblastomas usually present as a single, mildly painful, rapidly enlarging abdominal mass that arises in the right lobe of the liver in 55% to 60% of cases.[10] Rapid enlargement of these tumors rarely results in tumor rupture and hemorrhage. Tumors may reach up to 25 cm in size. Most tumors are solitary; however, up to 15% of tumors are multifocal. Some cases are associated with nonspecific symptoms such as weight loss, failure to thrive, or anorexia.[7] Significant elevations of α-fetoprotein are observed in 90% of patients, and rarely, a paraneoplastic syndrome can occur.
Evaluation
Ultrasonography and either computed tomography or magnetic resonance imaging are the imaging modalities used to define the extent of tumor involvement of the liver and aid in pre-surgical planning. A chest CT can help detect lung metastases; the lungs are the most common site of metastases, and up to 20% of cases present with metastases.[7] After imaging, a biopsy, α-fetoprotein level, liver function tests, and a hepatitis panel are performed as needed. Consent for participation in biologic studies should be requested before the biopsy is performed so the specimen can be properly allocated and handled.[9]
Treatment / Management
Surgical resection is the mainstay of treatment, with resectability of the tumor determining the need for neoadjuvant or adjuvant chemotherapy. At presentation, approximately 60% of tumors are unresectable.[11] If unresectable and chemotherapy fails to shrink the tumor to a resectable size, a liver transplant can be done and has a good long-term survival rate.[12] The benefit of radiation therapy remains unclear, though some unresectable cases respond well. α-Fetoprotein levels are useful for tracking surgical success and whether the tumor has metastasized.[10] An increased risk of posttransplant lymphoproliferative disorder after immunosuppression for liver transplant has been suggested in some publications.[6](B3)
Differential Diagnosis
The differential diagnosis includes hepatocellular carcinoma, focal nodular hyperplasia, hepatic adenoma, lymphoma, and metastases. Hepatocellular carcinoma can appear similar to the macrotrabecular subtype of hepatoblastoma; however, it usually affects an adult patient population with risk factors such as metabolic disorders, liver cirrhosis, or childhood hepatitis B infection. Focal nodular hyperplasia usually affects older children and adults. Hepatic adenoma can resemble pure fetal hepatoblastomas; however, these rarely occur in persons under 5 years of age unless they have an underlying metabolic disorder.
Staging
The Children’s Hepatic Tumors International Collaboration developed a staging and risk stratification system to standardize assessment of this tumor worldwide. This new staging system, called the Children’s Hepatic Tumors International Collaboration: Hepatoblastoma Stratification, incorporates confirmed prognostic factors from prior risk stratification systems, along with additional factors, to stratify patients into 4 risk groups.
The most predictive factors are α-fetoprotein levels, patient age, Pretreatment Extent of Disease (PRETEXT) group (I, II, III, or IV), the presence of metastases, and PRETEXT annotation factor. PRETEXT group is based on the extent of the tumor in the liver. PRETEXT annotation factor is determined to be positive if at least 1 of the following 5 factors are present: involvement of the vena cava or all 3 hepatic veins, or both (V); involvement of portal bifurcation or both right and left portal veins, or both (P); extrahepatic contiguous tumor extension (E); multifocal liver tumor (F); tumor rupture at diagnosis (R). Sex, low birth weight, prematurity, and Beckwith-Wiedemann syndrome were not found to be significant. Of note, the histologic type was not included in this risk stratification system but may be incorporated in the future. Validation of these risk groups is in progress.[2][13]
Prognosis
Prognosis is based on numerous factors, including age of diagnosis, PRETEXT group, metastases, α-fetoprotein levels, histologic subtype, completeness of resection, and clinical stage of the disease. With certainty, the well-differentiated fetal subtype is associated with a better prognosis than the small cell undifferentiated and macrotrabecular subtypes, which are associated with an unfavorable prognosis.[13] Of note, the histologic subtype is prognostically valuable only before chemotherapy. α-Fetoprotein is typically high at diagnosis, but a significant drop after neoadjuvant chemotherapy portends a better response to treatment. Younger age of diagnosis has historically been a poor prognostic factor; however, recent studies have called this into question, providing evidence that these younger patients do just as well as older children.[14] Specifically, children younger than 1 year of age have a better prognosis, and children older than 6 years of age have a worse prognosis.[2] Tumor presence at the resection margin, multifocality, and metastases have been shown to be poor prognostic factors. β-catenin expression has been associated with shorter event-free survival, while epithelial cell adhesion molecule (EpCAM) expression has been associated with higher tumor viability and poorer response to neoadjuvant chemotherapy.[15][16]
Complications
Complications include:
- Intraperitoneal tumor rupture
- Complications related to chemotherapy
- Posttransplant complications
- Psychosocial effects of treatment and painful procedures
Enhancing Healthcare Team Outcomes
Improved clinical outcomes are achieved when hepatoblastomas are managed by an interprofessional team that may include a pediatrician, oncologist, radiologist, pediatric surgeon, hepatologist, and a transplant specialist.[17] After surgical resection, these children are monitored by intensive care unit nurses, therapists, dietitians, and an intensivist.
Media
(Click Image to Enlarge)
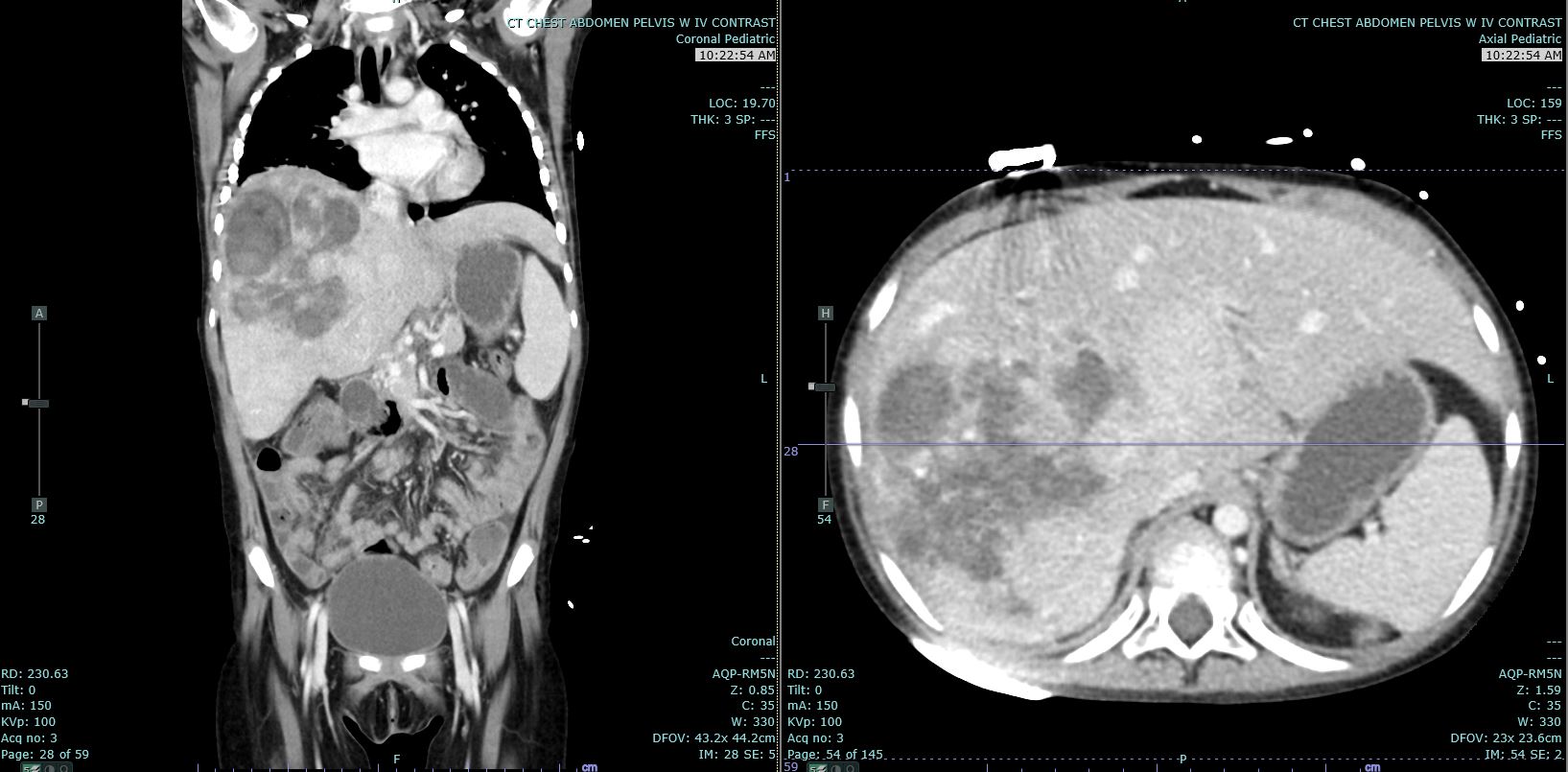
Hepatoblastoma Contributed by Melinda Smith MD, West Virginia University
(Click Image to Enlarge)
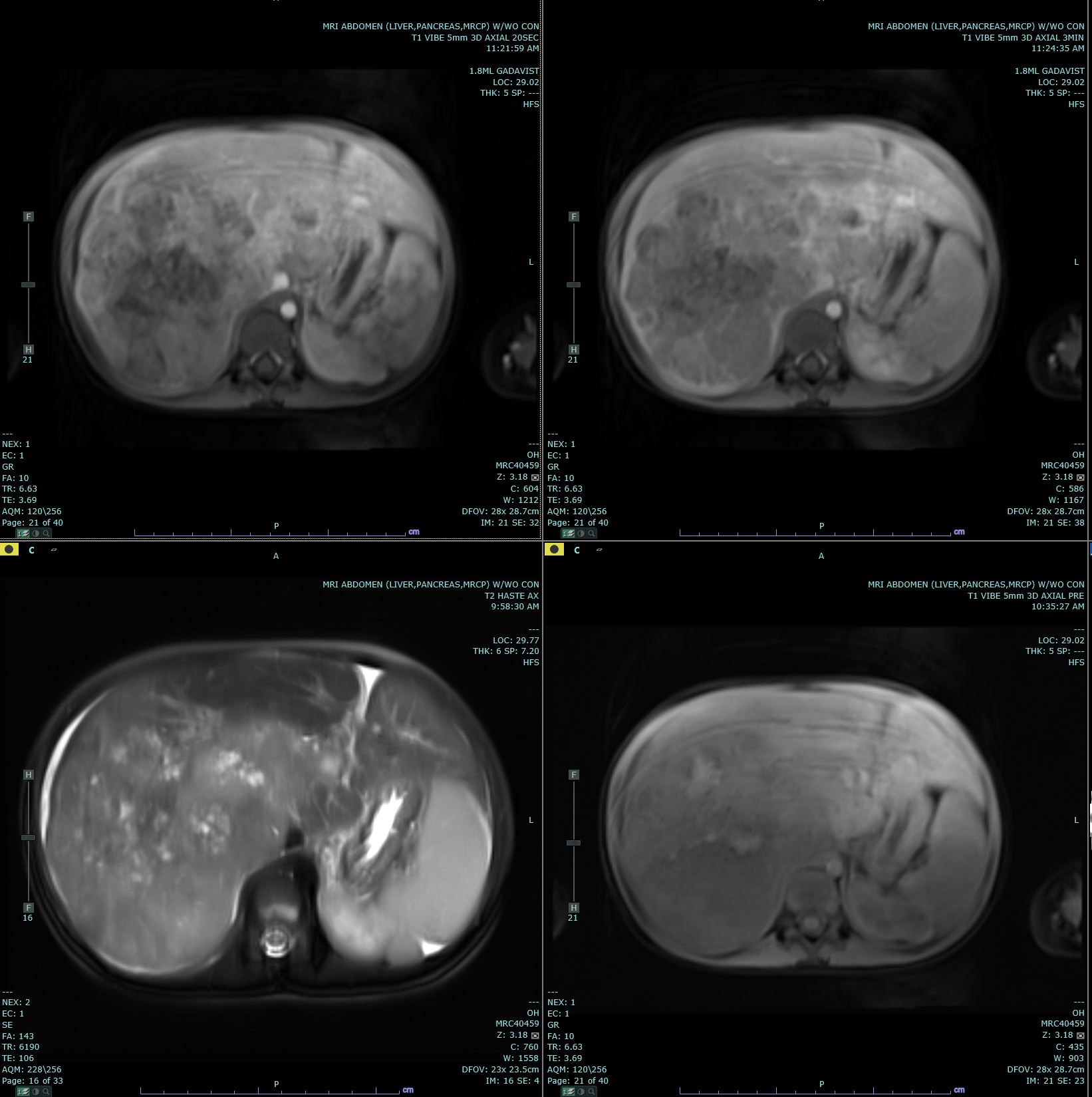
Hepatoblastoma Contributed by Melinda Smith, MD, West Virginia University
(Click Image to Enlarge)
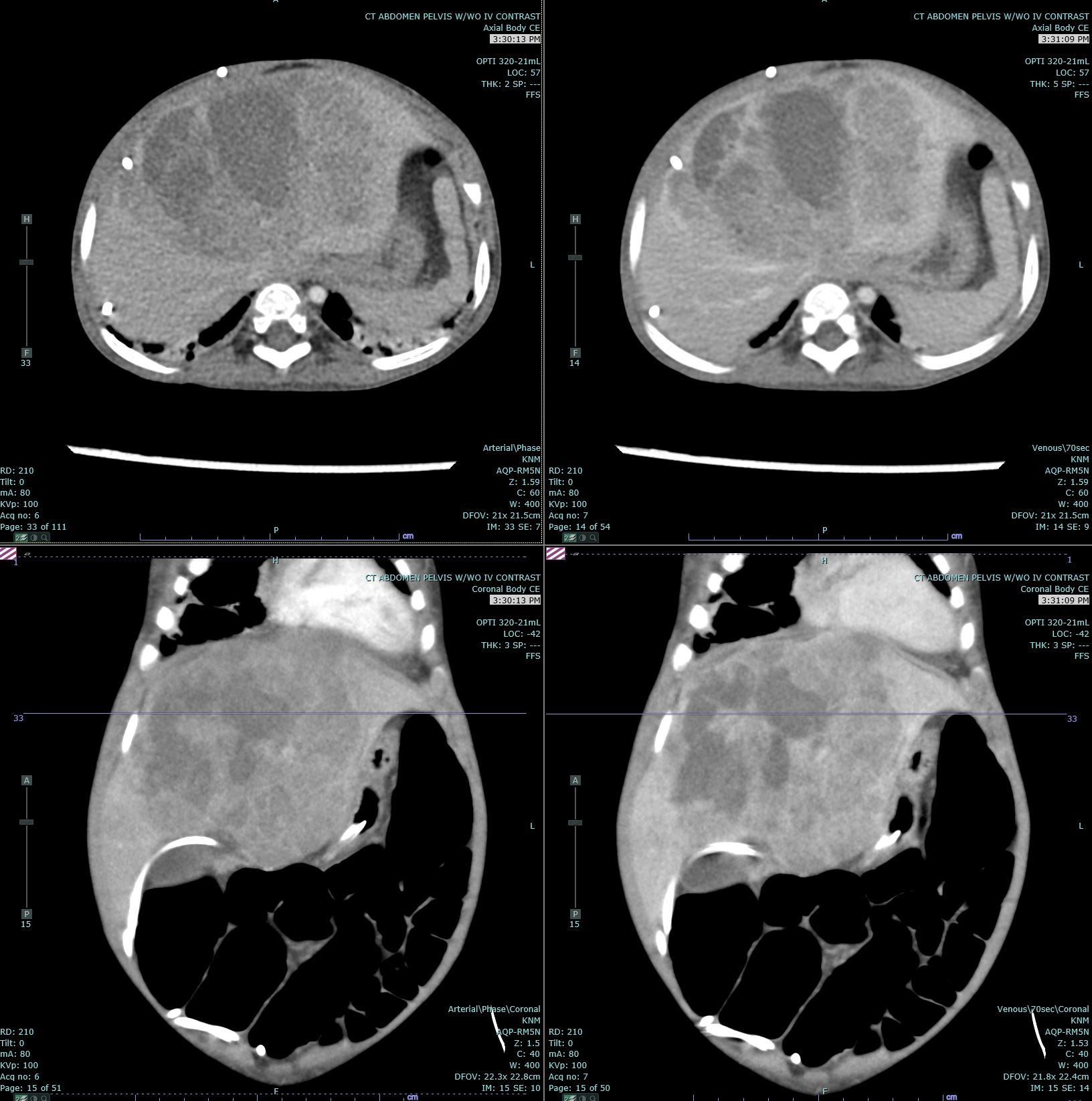
Hepatoblastoma Contributed by Melinda Smith, MD, West Virginia University
(Click Image to Enlarge)
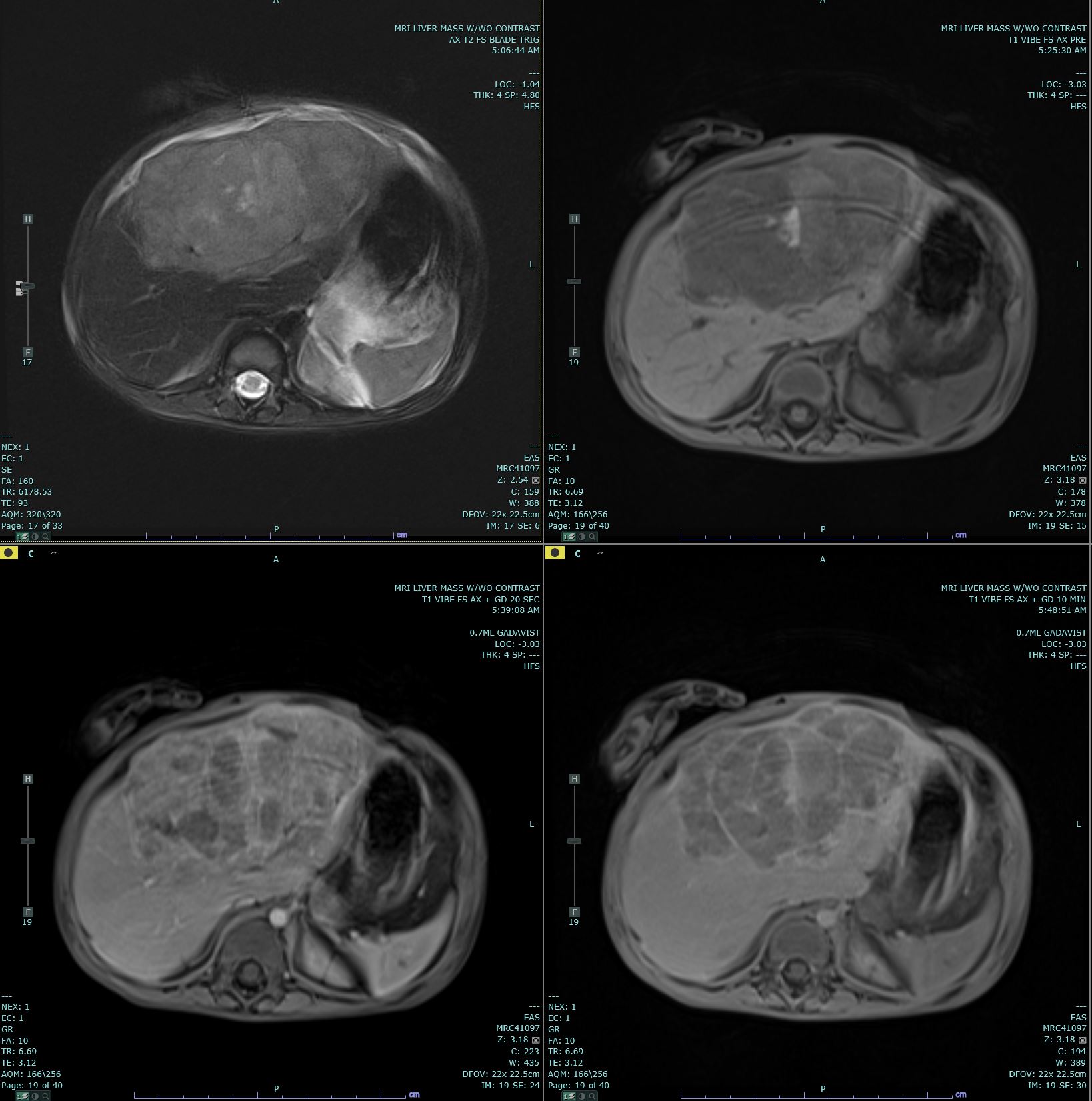
Hepatoblastoma Contributed by Melinda Smith, MD, West Virginia University
(Click Image to Enlarge)
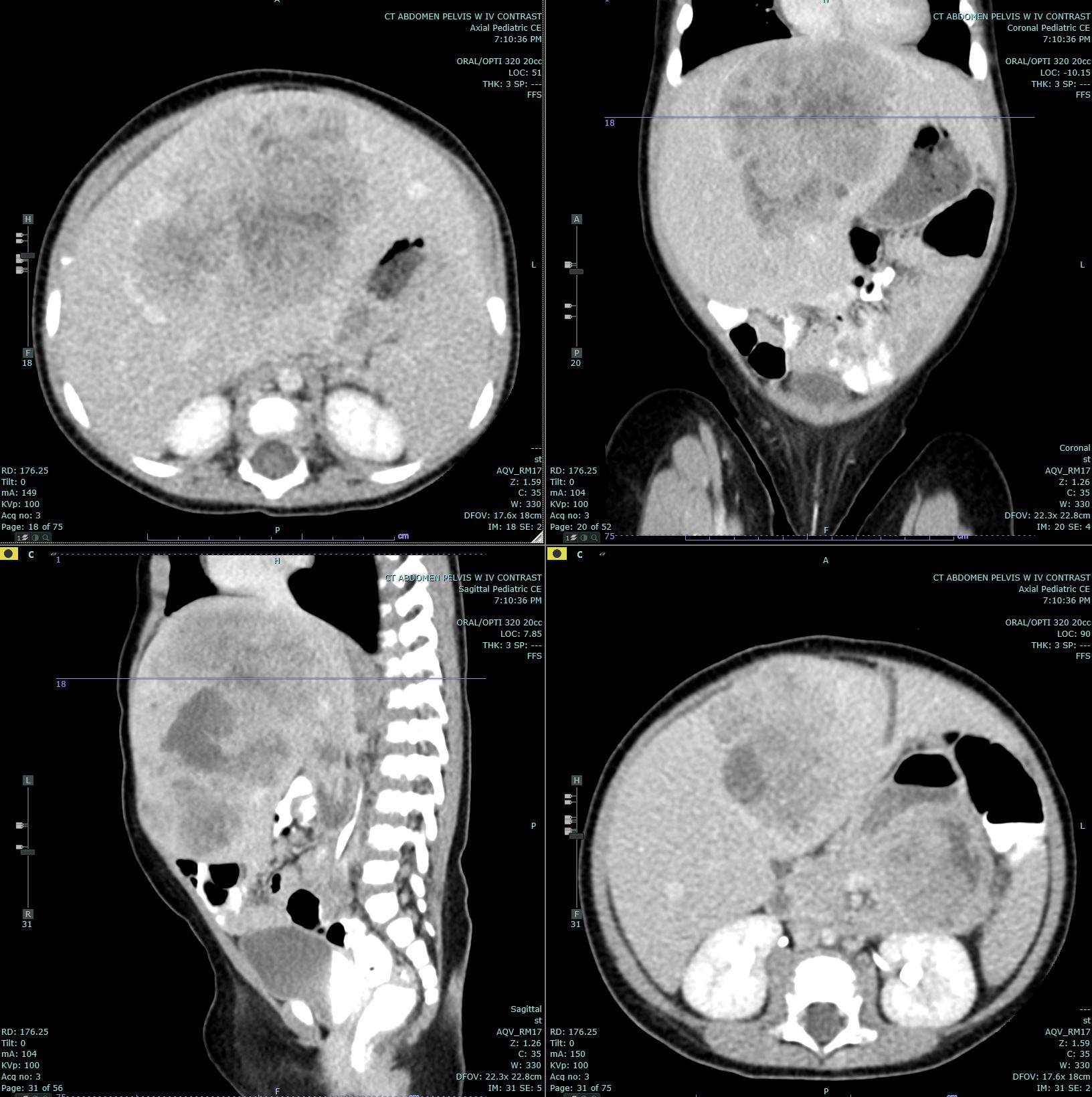
Hepatoblastoma Contributed by Melinda Smith, MD, West Virginia University
References
Sharma D, Subbarao G, Saxena R. Hepatoblastoma. Seminars in diagnostic pathology. 2017 Mar:34(2):192-200. doi: 10.1053/j.semdp.2016.12.015. Epub 2016 Dec 23 [PubMed PMID: 28126357]
Czauderna P, Lopez-Terrada D, Hiyama E, Häberle B, Malogolowkin MH, Meyers RL. Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Current opinion in pediatrics. 2014 Feb:26(1):19-28. doi: 10.1097/MOP.0000000000000046. Epub [PubMed PMID: 24322718]
Level 3 (low-level) evidenceFinegold MJ, Lopez-Terrada DH, Bowen J, Washington MK, Qualman SJ, College of American Pathologists. Protocol for the examination of specimens from pediatric patients with hepatoblastoma. Archives of pathology & laboratory medicine. 2007 Apr:131(4):520-9 [PubMed PMID: 17425379]
Heck JE, Meyers TJ, Lombardi C, Park AS, Cockburn M, Reynolds P, Ritz B. Case-control study of birth characteristics and the risk of hepatoblastoma. Cancer epidemiology. 2013 Aug:37(4):390-5. doi: 10.1016/j.canep.2013.03.004. Epub 2013 Apr 1 [PubMed PMID: 23558166]
Level 2 (mid-level) evidenceCuria MC, Zuckermann M, De Lellis L, Catalano T, Lattanzio R, Aceto G, Veschi S, Cama A, Otte JB, Piantelli M, Mariani-Costantini R, Cetta F, Battista P. Sporadic childhood hepatoblastomas show activation of beta-catenin, mismatch repair defects and p53 mutations. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2008 Jan:21(1):7-14 [PubMed PMID: 17962810]
Level 2 (mid-level) evidenceNg K, Rana A, Masand P, Patel K, Heczey A, Goss J, Himes R. Fatal Central Nervous System Post-Transplant Lymphoproliferative Disease in a Patient Who Underwent Liver Transplantation for Hepatoblastoma. Journal of pediatric gastroenterology and nutrition. 2018 Jan:66(1):e21-e23. doi: 10.1097/MPG.0000000000001725. Epub [PubMed PMID: 28837514]
Zhong S, Zhao Y, Fan C. Hepatoblastoma with pure fetal epithelial differentiation in a 10-year-old boy: A rare case report and review of the literature. Medicine. 2018 Jan:97(2):e9647. doi: 10.1097/MD.0000000000009647. Epub [PubMed PMID: 29480877]
Level 3 (low-level) evidencePateva IB, Egler RA, Stearns DS. Hepatoblastoma in an 11-year-old: Case report and a review of the literature. Medicine. 2017 Jan:96(2):e5858. doi: 10.1097/MD.0000000000005858. Epub [PubMed PMID: 28079820]
Level 3 (low-level) evidenceLópez-Terrada D, Alaggio R, de Dávila MT, Czauderna P, Hiyama E, Katzenstein H, Leuschner I, Malogolowkin M, Meyers R, Ranganathan S, Tanaka Y, Tomlinson G, Fabrè M, Zimmermann A, Finegold MJ, Children's Oncology Group Liver Tumor Committee. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2014 Mar:27(3):472-91. doi: 10.1038/modpathol.2013.80. Epub 2013 Sep 6 [PubMed PMID: 24008558]
Level 3 (low-level) evidenceHiyama E. Pediatric hepatoblastoma: diagnosis and treatment. Translational pediatrics. 2014 Oct:3(4):293-9. doi: 10.3978/j.issn.2224-4336.2014.09.01. Epub [PubMed PMID: 26835349]
Meyers RL, Tiao G, de Ville de Goyet J, Superina R, Aronson DC. Hepatoblastoma state of the art: pre-treatment extent of disease, surgical resection guidelines and the role of liver transplantation. Current opinion in pediatrics. 2014 Feb:26(1):29-36. doi: 10.1097/MOP.0000000000000042. Epub [PubMed PMID: 24362406]
Level 3 (low-level) evidenceUchida H, Sakamoto S, Sasaki K, Takeda M, Hirata Y, Fukuda A, Hishiki T, Irie R, Nakazawa A, Miyazaki O, Nosaka S, Kasahara M. Surgical treatment strategy for advanced hepatoblastoma: Resection versus transplantation. Pediatric blood & cancer. 2018 Dec:65(12):e27383. doi: 10.1002/pbc.27383. Epub 2018 Aug 7 [PubMed PMID: 30084209]
Meyers RL, Maibach R, Hiyama E, Häberle B, Krailo M, Rangaswami A, Aronson DC, Malogolowkin MH, Perilongo G, von Schweinitz D, Ansari M, Lopez-Terrada D, Tanaka Y, Alaggio R, Leuschner I, Hishiki T, Schmid I, Watanabe K, Yoshimura K, Feng Y, Rinaldi E, Saraceno D, Derosa M, Czauderna P. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. The Lancet. Oncology. 2017 Jan:18(1):122-131. doi: 10.1016/S1470-2045(16)30598-8. Epub 2016 Nov 22 [PubMed PMID: 27884679]
Dall'Igna P, Brugieres L, Christin AS, Maibach R, Casanova M, Alaggio R, de Goyet JV, Zsiros J, Morland B, Czauderna P, Childs M, Aronson DC, Branchereau S, Brock P, Perilongo G. Hepatoblastoma in children aged less than six months at diagnosis: A report from the SIOPEL group. Pediatric blood & cancer. 2018 Jan:65(1):. doi: 10.1002/pbc.26791. Epub 2017 Sep 18 [PubMed PMID: 28921839]
Kiruthiga KG, Ramakrishna B, Saha S, Sen S. Histological and immunohistochemical study of hepatoblastoma: correlation with tumour behaviour and survival. Journal of gastrointestinal oncology. 2018 Apr:9(2):326-337. doi: 10.21037/jgo.2018.01.08. Epub [PubMed PMID: 29755772]
Wu JF, Chang HH, Lu MY, Jou ST, Chang KC, Ni YH, Chang MH. Prognostic roles of pathology markers immunoexpression and clinical parameters in Hepatoblastoma. Journal of biomedical science. 2017 Aug 29:24(1):62. doi: 10.1186/s12929-017-0369-1. Epub 2017 Aug 29 [PubMed PMID: 28851352]
Shanmugam N, Scott JX, Kumar V, Vij M, Ramachandran P, Narasimhan G, Reddy MS, Kota V, Munirathnam D, Kelgeri C, Sundaram K, Rela M. Multidisciplinary management of hepatoblastoma in children: Experience from a developing country. Pediatric blood & cancer. 2017 Mar:64(3):. doi: 10.1002/pbc.26249. Epub 2016 Oct 26 [PubMed PMID: 27781375]