Introduction
Immunoglobulin A vasculitis (IgAV), formerly Henoch-Schönlein purpura, involves the small vessels of the joints, kidneys, gastrointestinal tract, and skin. IgAV can also affect the central nervous system and the lungs; however, these findings are rare. IgAV is an acute IgA-mediated disorder that is typically self-limited and treated with supportive care; however, serious complications, such as renal failure, can occur.
Henoch-Schönlein purpura is named after the German physician Johann Schönlein and his student Eduard Henoch. Schönlein identified the association between joint pain and purpura, and Henoch identified gastrointestinal tract and renal involvement. Although Henoch-Schönlein purpura is named after Henoch and Schönlein, an English physician, William Heberden, was the first to describe the disorder in the early 1800s.[1] IgA vasculitis is now the preferred term because of a tendency toward etiology-based rather than eponym-based nomenclature.
IgA vasculitis with nephritis shares many features with IgA nephropathy, the most common glomerulonephritis worldwide. The primary differences are that IgAV with nephritis is more likely to present in children younger than 15 years, whereas IgA nephropathy usually presents in patients older than 15 years. IgAV with nephritis is more likely to present with extrarenal symptoms, whereas IgA nephropathy presents more often with gross hematuria. Histologic findings in IgAV with nephritis show more capillary staining and glomerular injury than findings in IgA nephropathy.
IgAV with nephritis has a clinical remission rate of 98%; comparatively, 30% to 50% of patients with IgA nephropathy progress to end-stage renal disease within 20 years of diagnosis.[2] Another interesting difference is that IgAV with nephritis presents more often during the winter cold season, while IgA nephropathy does not show seasonality. Much of the difference between the 2 disease processes results from IgAV being predominantly a disease of children, whereas IgA nephropathy is primarily a disease of adults.[2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Environmental, genetic, and antigenic factors appear to contribute to the etiology of IgA vasculitis. Many patients report a preceding infection. Upper respiratory tract infections are the most common; however, patients may also present with an antecedent gastrointestinal tract or pharyngeal infection. Group A streptococcal infections have been detected in cultures from more than 30% of patients with IgAV and nephritis.[4][5] Other causative agents include:
- Coxsackie virus
- Hepatitis A and hepatitis B vaccines (and hepatitis vaccines)
- Mycoplasma spp
- Parvovirus B19
- Infectious mononucleosis
- Subacute bacterial endocarditis
- Helicobacter pylori infection
- Yersinia and Shigella spp
- Salmonella spp
- Brucellosis spp
- Legionella spp
- Campylobacter spp
- Varicella
- Parainfluenza virus
- Influenza virus (and vaccine)
- Respiratory syncytial virus
- Rotavirus
- Cytomegalovirus (CMV) reactivation
- Adenoviruses [4]
More recently, IgAV has also been found to be associated with COVID-19 infections.[5][6][7] The virus directly damages blood vessels, leading to inflammation and immune complex formation. IgAV has also been associated with COVID-19 immunizations.[8][9] Furthermore, several genetic associations have been linked to Henoch-Schönlein purpura.[10][11] Human leukocyte antigen (HLA) class 2 associations include HLA-DQA1*01:01, HLA-DQB1*05:01, and HLA-DRB1*01:01; 3 alleles, HLA-DRB1*04, HLA-DRB1*16, and HLA-DRB1*16:02, were highly related. Mediterranean fever gene (MEFV) variants are also related, especially E148Q and M694V, with E148Q being the most common subtype. Exon 10 mutations are associated with particularly severe symptoms. The AG genotype of the interleukin 1β (IL1B) gene is associated with arthritis. The rs10818488 locus in the tumor necrosis factor receptor superfamily member 1A (TNFRSF1A) gene may reflect the severity of kidney involvement. Glutathione S-transferase mu 1 (GSTM1) and glutathione S-transferase pi 1 (GSTP1) gene polymorphisms may be associated with gastrointestinal and urinary complications. Urinary protein analysis carries an association; these proteins are P09417, Q725L0, P60900, P14550, and P09668. Finally, skin involvement in IgA vasculitis can show expression of miRNA-155-5p, miRNA-223-3p, and let-7b.
Epidemiology
IgAV is a rare disorder that typically affects children; however, the condition can also be seen in adults and adolescents. Most affected children are younger than 10 years. IgAV is often more severe and more likely to cause long-term renal disease in adults.[4] IgAV is the most common vasculitis among children, affecting 10 to 20 per 100,000 children per year.[12] IgAV is slightly more common among boys than girls but has an approximately equal sex distribution in adults.[13]
Pathophysiology
The pathophysiology of IgA vasculitis is not fully understood; however, IgA plays a significant role. The mucous membranes of the salivary glands, lungs, and gastrointestinal tract produce IgA. Plasma B cells produce other classes of immunoglobulins (IgG, IgE, and IgM).[14] IgA1 is implicated in IgAV rather than the IgA2 subtype and is associated with abnormally low galactose levels.[8]
IgA-antibody immune complexes are formed in response to antigenic exposure from an infection or medication. These immune complexes are then deposited in the small vessels, usually capillaries, of the skin, joints, kidneys, and gastrointestinal tract. IgA-antibody immune complex deposition results in an influx of inflammatory mediators, such as prostaglandins. The complement system can also be activated when C3-receptor lymphocytes bind to immune complexes that deposit on vessel walls, contributing to the hyperinflammatory response. Immune complex deposition in the intestinal wall may cause gastrointestinal tract hemorrhage.[4] Immune complexes deposited in the skin cause palpable purpura and petechiae.[8]
Histopathology
Renal biopsy is the most definitive tool for diagnosing both IgAV with nephritis and IgA nephropathy. Both are likely to show mesangial IgA deposits. IgAV with nephritis is more likely to show capillary and subendothelial IgA deposits, as well as neutrophilic infiltration. Renal deposits of IgA-mediated immune complexes may result in mild proliferative or severe crescentic glomerulonephritis.[2][4] IgAV with nephritis is also more likely to show mixed immune complexes with IgA and IgG components, while biopsies from patients diagnosed with IgA nephropathy tend to show only IgA complexes.
History and Physical
The classic presentation of IgA vasculitis includes palpable purpura, gastrointestinal complaints, arthralgias, and renal involvement. Patients may present with any of the following signs or symptoms:
- Rash
- Fatigue
- Headache
- Fever
- Joint pain
- Subcutaneous edema
- Diarrhea
- Hematemesis
- Abdominal pain
- Vomiting
- Rectal bleeding
- Scrotal edema
Physical Examination
Skin
Skin involvement is present in all patients with IgAV and is usually the earliest sign.[1] The rash associated with IgA vasculitis often starts as erythematous, macular, or urticarial lesions (see Image. Henoch-Schönlein Purpura). The rash then develops into palpable purpura and petechiae that most commonly affect the buttocks and lower extremities, particularly the extensor surfaces. Approximately one-third of patients experience the rash on the upper extremities and trunk, but the findings are primarily in dependent structures, which vary by age group and mobility status.[12] The lesions can become bullous or necrotic, and the bullous form has been associated with progressive, treatment-resistant renal failure.[15] The lesions change from red to purple, then rust-colored, before fading. These changes occur over approximately 10 days.[4]
Gastrointestinal Tract
Gastrointestinal tract findings may occur in 10% to 40% of patients before the rash.[12] Patients may present with nausea and vomiting, which may worsen after meals. Potentially life-threatening complications include intussusception, bowel perforation, bowel gangrene, and massive hemorrhage. Intussusception is the most common life-threatening gastrointestinal tract complication, affecting 3% to 4% of patients with IgAV.[1] Other presenting signs and symptoms include extensive skin lesions, an increased D-dimer level, atypical rashes, and joint involvement.[16]
Renal
Renal symptoms typically occur within 1 to 3 months after the development of a rash in 20% to 55% of children with IgAV.[12] Renal manifestations include hematuria, proteinuria, nephrotic syndrome, nephritic syndrome, and renal failure. The most common renal manifestation is microscopic hematuria.[12] Severe proteinuria may present as nephrotic syndrome, and patients with persistent proteinuria are at high risk of developing progressive glomerulonephritis. Patients may also develop ureteric obstructions. Approximately 50% of patients develop renal manifestations, with less than 1% progressing to end-stage renal failure.[17] Death from IgA vasculitis is rare; however, renal disease is the most common cause of morbidity and mortality in patients with the disorder.[4]
Joints
Approximately 15% of patients with IgA vasculitis present with arthritis as the initial symptom, and overall, arthralgia or arthritis occurs in 75% of children with the disorder.[12] Patients often present with painful, swollen joints, most commonly in the knees, ankles, hands, and feet. The arthralgias are typically transient and nondestructive
Central Nervous System
Central nervous system involvement is rare; however, when it occurs, patients may present with headaches, dizziness, ataxia, seizures, irritability, mononeuropathy, intracranial hemorrhage, or acute motor sensory axonal neuropathy.[12]
Evaluation
The diagnosis of IgA vasculitis is made based on the presence of petechiae without thrombocytopenia or palpable purpura that predominantly affects the lower extremities, plus at least 1 of the following 4 characteristics:
- Abdominal pain
- Arthralgia or arthritis
- Renal involvement (proteinuria, red blood cell casts, or hematuria)
- Proliferative glomerulonephritis or leukocytoclastic vasculitis with predominant deposition of IgA on histology [1]
Clinicians should order a urinalysis with microscopy to identify hematuria, proteinuria, or red blood cell casts. If the urine dipstick results are positive for protein, a 24-hour collection should be obtained to quantify the protein excretion.[1] A positive urine protein result is considered a harbinger of IgAV recurrence.[18] Measurement of serum IgA levels is nondiagnostic because levels vary significantly, but a significant elevation is suggestive of the disease. Antistreptolysin O titers should also be obtained. Furthermore, ultrasonography is often an initial imaging test to rule out hydronephrosis. Endoscopy can show purpura in the duodenum, stomach, and colon. Elevated D-dimer levels are seen with intussusception.[19]
Treatment / Management
Unless renal involvement is present, symptomatic and supportive care are the foundations of treatment for patients with IgAV. Acetaminophen or opioids are often preferred for pain control over nonsteroidal anti-inflammatory drugs in the setting of gastrointestinal tract or renal involvement.[20] Severe abdominal pain should prompt consideration of prednisone or prednisolone with a taper.[21] Ultrasonography confirms intussusception, and air-contrast enemas or surgical correction may follow.[19][22](B2)
Supportive and Symptomatic Care
- Rehydration with intravenous fluids
- Pain control
- Wound care for ulcerative skin lesions
Management of IgA Vasculitis Nephritis
Angiotensin-converting enzyme inhibitors: Angiotensin-converting enzyme inhibition can decrease proteinuria.[23] These agents are described as renoprotective because they decrease glomerular cell proliferation and fibrosis. However, angiotensin-converting enzyme inhibitors also have the propensity to induce a rare adult form of this disease.[24][25](B3)
Inhibition of the renin-angiotensin-aldosterone system has been shown to reduce proteinuria across various pathologies. Reducing long-term renal complications, such as decreased glomerular filtration rate, chronic kidney disease, and progression to end-stage renal disease, is critical. These agents are most often used in adults, who have less spontaneous recovery than children and are more likely to have hypertension.
Plasmapheresis or plasma exchange may be used concurrently with immunosuppressive therapy. Plasmapheresis decreases inflammation and inhibits glomerular crescent formation by clearing IgA complexes.[11] The addition of corticosteroids improves disease resolution.(B3)
Immunosuppressants: A significant number are applicable in this disease. Azathioprine has been used successfully in recurrent disease, particularly with skin involvement. Results from studies showed that methotrexate had efficacy in patients with severe and recurrent gastrointestinal tract involvement. Imipramine, when combined with corticosteroids, has shown good effects, including symptomatic relief and a reduction in proteinuria. Results from studies showed that tacrolimus improved renal function. Mycophenolate mofetil is particularly helpful in patients with recurrent renal and gastrointestinal tract involvement. Cyclophosphamide, often used in combination with other immunosuppressants, has demonstrated utility in children.
Corticosteroids: Early oral prednisone treatment is helpful in treating renal, joint, and gastrointestinal tract manifestations. Prednisone does not prevent renal disease but reduces the risk of developing persistent renal complications in children.[4] Results from several randomized controlled trials suggested that prednisone minimizes the duration and severity of abdominal pain during the first 2 weeks of treatment.[20] A recent algorithm recommends that oral prednisolone be given for low-level vasculitis nephritis, oral or pulse corticosteroids for moderate disease, and oral or pulse corticosteroids with intravenous cyclophosphamide for severe forms.[10] For second-line therapy, rituximab, azathioprine, mycophenolate mofetil, corticosteroids, and cyclophosphamide are advocated.(B3)
Fish oil, when administered in a standardized pharmaceutical formulation, has been shown in several trials to be more effective than a placebo for IgA nephropathy through potential anti-inflammatory mechanisms and may also be effective in IgAV.
Intravenous immunoglobulin can also be effective in patients with rapidly progressive glomerulonephritis. Mechanisms of action include impairing autoreactive T cells by blocking binding to antigen-presenting cells, downregulating antibody production by B cells, and blocking the Fc receptor–mediated immune response.[26] (A1)
Dapsone suppresses interleukin-8, myeloperoxidase, and leukocyte chemotactic activity, collectively producing an anti-inflammatory effect.[27] Dapsone has notable effects on cutaneous vasculitis and gastrointestinal tract involvement, but significantly fewer effects on nephritic disease.[10][11](B3)
Differential Diagnosis
The differential diagnosis of IgAV includes the following:
- IgA nephropathy
- Acute kidney injury
- Acute glomerulonephritis
- Immune thrombocytopenia
- Disseminated intravascular coagulation
- Thrombotic thrombocytopenic purpura
- Hemolytic uremic syndrome
- Meningococcal meningitis
- Hypersensitivity vasculitis
- Systemic lupus erythematosus
- Polyarteritis nodosa
- Bacterial endocarditis
- Inflammatory bowel disease
- Granulomatosis with polyangiitis
- Rocky Mountain spotted fever
Prognosis
IgA vasculitis is typically a self-limited illness that has an excellent prognosis in patients without renal involvement. Most patients fully recover in 4 weeks.[4] IgA vasculitis recurs in approximately one-third of patients within 4 to 6 months after the initial onset. The long-term morbidity of IgA vasculitis is dependent on the extent of renal involvement. Approximately 1% of patients with IgA vasculitis will develop end-stage renal disease and require a renal transplant.[20]
Complications
IgA vasculitis involves multiple organ systems, and the potential complications are relatively extensive. Potential complications include the following:
- Renal failure
- Proteinuria
- Hematuria
- Nephrotic syndrome
- Intussusception
- Gastrointestinal tract bleeding
- Bowel infarction
- Bowel perforation
- Central nervous system bleeding
- Seizures
- Neuropathy
- Pleural effusion
- Pulmonary hemorrhage
- Testicular torsion
Consultations
Patients who present with nephritic syndrome, nephrotic syndrome, hematuria, or rapidly worsening proteinuria should be urgently referred to a pediatric nephrologist.[20]
Deterrence and Patient Education
Patients should be educated that the symptoms will likely resolve within weeks but may recur. Although severe renal involvement is rare, patients with evidence of severe renal involvement require aggressive treatment and care by a nephrologist.
Pearls and Other Issues
Key facts regarding IgAV are as follows:
- IgAV is primarily a disease of children, with only 10% of cases occurring in adults older than 18 years.
- Relapse is common in the first 4 to 6 months, but full recovery usually occurs with a mortality rate of less than 1%.
- The presentation can be similar to IgA nephropathy but with more skin, gastrointestinal tract, and joint manifestations and fewer chronic effects.
- Histology shows more IgA deposits in the capillaries and subendothelial space, and a more likely copresence of IgG than IgA nephropathy.
- Galactose-deficient IgA1 is commonly found in IgAV, especially in cases with renal involvement.
Enhancing Healthcare Team Outcomes
IgA vasculitis is typically a self-limited illness; however, patients may develop life-threatening complications such as intussusception, massive gastrointestinal tract hemorrhage, and renal failure. An interprofessional approach is necessary to adequately diagnose and treat the illness. Patients may present with nonspecific symptoms such as malaise, upper respiratory tract symptoms, or arthralgias before developing the characteristic rash. Nonspecific symptoms may delay diagnosis. Clinicians may see patients at different stages of the disease; therefore, medical staff must communicate and remain aware of potential complications. A surgical team and radiologist may be required to diagnose and treat intussusception.
An essential aspect of the disease process is adequate follow-up with frequent urinalyses to screen for potential renal involvement. Patients treated with corticosteroids may require assistance from pharmacists regarding adequate therapeutic dosing and tapering. Patients with severe renal disease will need to see a nephrology team comprising medical assistants, nurses, clinicians, and, rarely, a transplant team.
Media
(Click Image to Enlarge)
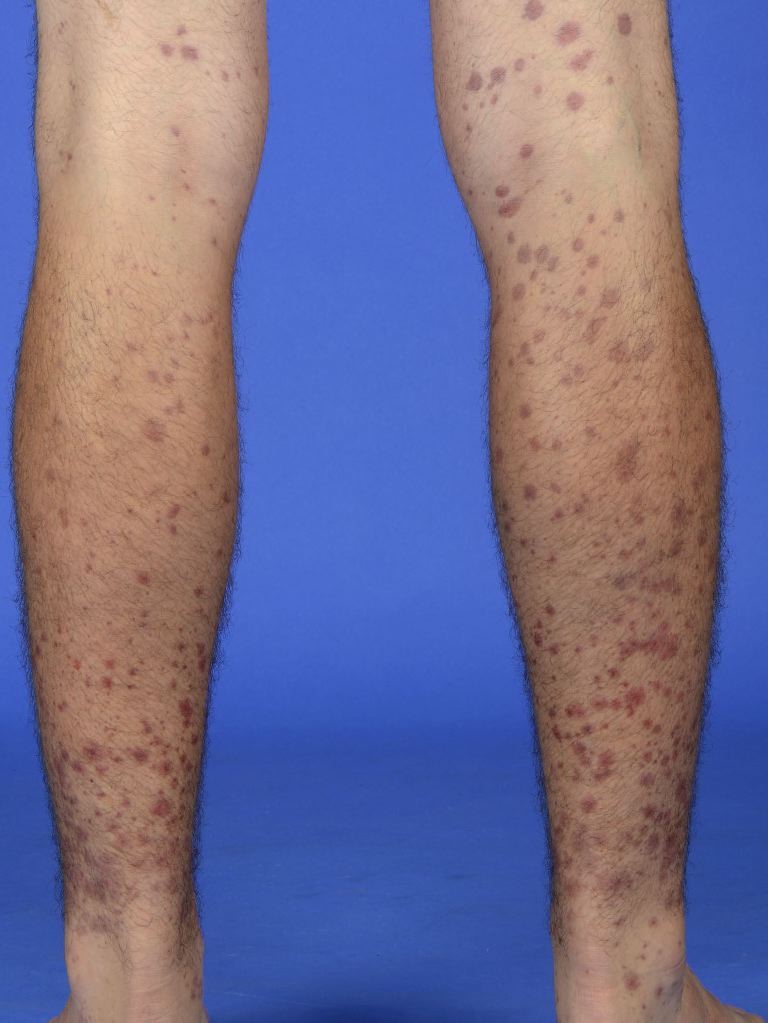
Henoch-Schönlein Purpura. Symmetrical distribution of palpable purpura and erythematous macules on the lower extremities, characteristic of the cutaneous presentation of IgA vasculitis.
References
Trnka P. Henoch-Schönlein purpura in children. Journal of paediatrics and child health. 2013 Dec:49(12):995-1003. doi: 10.1111/jpc.12403. Epub 2013 Oct 18 [PubMed PMID: 24134307]
Rajasekaran A, Julian BA, Rizk DV. IgA Nephropathy: An Interesting Autoimmune Kidney Disease. The American journal of the medical sciences. 2021 Feb:361(2):176-194. doi: 10.1016/j.amjms.2020.10.003. Epub 2020 Oct 8 [PubMed PMID: 33309134]
Selvaskandan H, Shi S, Twaij S, Cheung CK, Barratt J. Monitoring Immune Responses in IgA Nephropathy: Biomarkers to Guide Management. Frontiers in immunology. 2020:11():572754. doi: 10.3389/fimmu.2020.572754. Epub 2020 Oct 6 [PubMed PMID: 33123151]
Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. American family physician. 2009 Oct 1:80(7):697-704 [PubMed PMID: 19817340]
Nikolaishvili M, Pazhava A, Di Lernia V. Viral Infections May Be Associated with Henoch-Schönlein Purpura. Journal of clinical medicine. 2023 Jan 16:12(2):. doi: 10.3390/jcm12020697. Epub 2023 Jan 16 [PubMed PMID: 36675626]
Salem Y, Alam Z, Shalabi MM, Hosler GA, Acharya S. IgA Vasculitis Associated With COVID-19. Cureus. 2023 May:15(5):e38725. doi: 10.7759/cureus.38725. Epub 2023 May 8 [PubMed PMID: 37292558]
Lee MO, Yoo SJ. Henoch-Schönlein purpura following mRNA COVID-19 vaccination: a case report. Clinical and experimental vaccine research. 2024 Apr:13(2):166-170. doi: 10.7774/cevr.2024.13.2.166. Epub 2024 Apr 30 [PubMed PMID: 38752010]
Level 3 (low-level) evidenceXu L, Li Y, Wu X. IgA vasculitis update: Epidemiology, pathogenesis, and biomarkers. Frontiers in immunology. 2022:13():921864. doi: 10.3389/fimmu.2022.921864. Epub 2022 Oct 3 [PubMed PMID: 36263029]
Jin L, Zhao C, Xiong J, Chang Q, Su Y, Dou B, Zhang L, He P. The impact of COVID-19 on recovery in Henoch-Schönlein purpura patients: a cross-sectional questionnaire study during a distinctive period. Frontiers in pediatrics. 2025:13():1635822. doi: 10.3389/fped.2025.1635822. Epub 2026 Jan 16 [PubMed PMID: 41624826]
Level 2 (mid-level) evidenceSestan M, Jelusic M. Diagnostic and Management Strategies of IgA Vasculitis Nephritis/Henoch-Schönlein Purpura Nephritis in Pediatric Patients: Current Perspectives. Pediatric health, medicine and therapeutics. 2023:14():89-98. doi: 10.2147/PHMT.S379862. Epub 2023 Mar 7 [PubMed PMID: 36915829]
Level 3 (low-level) evidenceXu M, Wei Z, Wang L, Liang W, Gao F, Sang S, Zhang R. Advances in multi-omics and therapeutic studies of Henoch-Schönlein purpura. Italian journal of pediatrics. 2025 Dec 5:52(1):5. doi: 10.1186/s13052-025-02174-6. Epub 2025 Dec 5 [PubMed PMID: 41350890]
Level 3 (low-level) evidenceHetland LE, Susrud KS, Lindahl KH, Bygum A. Henoch-Schönlein Purpura: A Literature Review. Acta dermato-venereologica. 2017 Nov 15:97(10):1160-1166. doi: 10.2340/00015555-2733. Epub [PubMed PMID: 28654132]
Lei WT, Tsai PL, Chu SH, Kao YH, Lin CY, Fang LC, Shyur SD, Lin YW, Wu SI. Incidence and risk factors for recurrent Henoch-Schönlein purpura in children from a 16-year nationwide database. Pediatric rheumatology online journal. 2018 Apr 16:16(1):25. doi: 10.1186/s12969-018-0247-8. Epub 2018 Apr 16 [PubMed PMID: 29661187]
Sugino H, Sawada Y, Nakamura M. IgA Vasculitis: Etiology, Treatment, Biomarkers and Epigenetic Changes. International journal of molecular sciences. 2021 Jul 14:22(14):. doi: 10.3390/ijms22147538. Epub 2021 Jul 14 [PubMed PMID: 34299162]
Popov H, Koleva T, Stoyanov GS. Bullous Henoch-Schönlein Purpura and Associated Nephritis: A Pathological Case Report. Cureus. 2023 Feb:15(2):e35051. doi: 10.7759/cureus.35051. Epub 2023 Feb 16 [PubMed PMID: 36942172]
Level 3 (low-level) evidenceGuo A, Wang Y, Nan X, Zhang Y, Liu P, Tu C. Clinical review of adult Henoch-Schönlein purpura and analysis of predictors related to gastrointestinal involvement. Frontiers in immunology. 2025:16():1694532. doi: 10.3389/fimmu.2025.1694532. Epub 2025 Dec 5 [PubMed PMID: 41425593]
Guo D, Lam JM. Henoch-Schönlein purpura. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne. 2016 Oct 18:188(15):E393. doi: 10.1503/cmaj.151072. Epub 2016 Jul 18 [PubMed PMID: 27431304]
Cao T, Yang HM, Huang J, Hu Y. Risk factors associated with recurrence of Henoch-Schonlein purpura: a retrospective study. Frontiers in pediatrics. 2023:11():1164099. doi: 10.3389/fped.2023.1164099. Epub 2023 Jun 12 [PubMed PMID: 37377759]
Level 2 (mid-level) evidenceZhao Q, Yang Y, He SW, Wang XT, Liu C. Risk factors for intussusception in children with Henoch-Schönlein purpura: A case-control study. World journal of clinical cases. 2021 Aug 6:9(22):6244-6253. doi: 10.12998/wjcc.v9.i22.6244. Epub [PubMed PMID: 34434991]
Level 2 (mid-level) evidenceBluman J, Goldman RD. Henoch-Schönlein purpura in children: limited benefit of corticosteroids. Canadian family physician Medecin de famille canadien. 2014 Nov:60(11):1007-10 [PubMed PMID: 25551129]
Bista S, Adhikari Y, Karmacharya S, Joshi S, Pandey S, Adhikari N. Henoch-Schönlein purpura with antecedent allergic diseases in a 4-year-old child: a case report. Annals of medicine and surgery (2012). 2023 Jun:85(6):3066-3069. doi: 10.1097/MS9.0000000000000782. Epub 2023 May 8 [PubMed PMID: 37363590]
Level 3 (low-level) evidenceCui XH, Liu H, Fu L, Zhang C, Wang XD. Henoch-Schönlein purpura with intussusception and hematochezia in an adult: A case report. Medicine. 2019 Sep:98(36):e16981. doi: 10.1097/MD.0000000000016981. Epub [PubMed PMID: 31490379]
Level 3 (low-level) evidenceTudorache E, Azema C, Hogan J, Wannous H, Aoun B, Decramer S, Deschênes G, Ulinski T. Even mild cases of paediatric Henoch-Schönlein purpura nephritis show significant long-term proteinuria. Acta paediatrica (Oslo, Norway : 1992). 2015 Aug:104(8):843-8. doi: 10.1111/apa.12723. Epub 2014 Jul 29 [PubMed PMID: 24946692]
Level 3 (low-level) evidenceGonçalves R, Cortez Pinto H, Serejo F, Ramalho F. Adult Schönlein-Henoch purpura after enalapril. Journal of internal medicine. 1998 Oct:244(4):356-7 [PubMed PMID: 9797501]
Santos S, Santos F, Fróis Cunha I, Rato M, Camões S. An Unusual Complication of Lisinopril: A Report of Iatrogenic Henoch-Schönlein Purpura. Cureus. 2024 Aug:16(8):e68323. doi: 10.7759/cureus.68323. Epub 2024 Aug 31 [PubMed PMID: 39350862]
Nguyen B, Acharya C, Tangpanithandee S, Miao J, Krisanapan P, Thongprayoon C, Amir O, Mao MA, Cheungpasitporn W, Acharya PC. Efficacy and Safety of Plasma Exchange as an Adjunctive Therapy for Rapidly Progressive IgA Nephropathy and Henoch-Schönlein Purpura Nephritis: A Systematic Review. International journal of molecular sciences. 2023 Feb 16:24(4):. doi: 10.3390/ijms24043977. Epub 2023 Feb 16 [PubMed PMID: 36835388]
Level 1 (high-level) evidenceLee KH, Park JH, Kim DH, Hwang J, Lee G, Hyun JS, Heo ST, Choi JH, Kim M, Kim M, Kim SI, Eisenhut M, Kronbichler A, Shin JI. Dapsone as a potential treatment option for Henoch-Schönlein Purpura (HSP). Medical hypotheses. 2017 Oct:108():42-45. doi: 10.1016/j.mehy.2017.07.018. Epub 2017 Jul 17 [PubMed PMID: 29055398]