Introduction
Heme is an iron-containing porphyrin, specifically ferrous (Fe2+) protoporphyrin IX, serving as an essential prosthetic group for various proteins involved in oxygen transport, redox reactions, and cellular metabolism (see Image. Chemical Structure of Heme A).[1] As a prosthetic group, heme plays a critical role in hemoprotein function, most notably hemoglobin and myoglobin, enabling reversible oxygen binding. Additionally, heme functions as an indispensable cofactor for numerous enzymes, including cytochromes of the electron transport chain (ETC), cytochrome P450 (CYP450) enzymes, catalase, and nitric oxide synthase, thereby contributing to key biochemical and metabolic pathways, such as oxidative phosphorylation, detoxification, and reactive oxygen species metabolism.
Heme synthesis is a highly conserved, multistep process occurring in all living cells, with maximal activity in 2 major tissues: erythroid precursors in the bone marrow, where heme production supports hemoglobin synthesis, and hepatocytes in the liver, the primary site of heme utilization for CYP450-mediated metabolic processes. At the cellular level, heme biosynthesis consists of an 8-step enzymatic pathway distributed between the mitochondria and cytosol, beginning with glycine and succinyl coenzyme A (succinyl-CoA) and culminating in the insertion of ferrous iron into protoporphyrin IX. Tight regulatory control of this pathway maintains balance between heme production and cellular demand, as both heme deficiency and excess exert cytotoxic effects. Clinically, defects in heme synthesis produce a spectrum of disorders, including porphyrias and sideroblastic anemias, underscoring the importance of this pathway in human physiology and disease.[2]
Fundamentals
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Fundamentals
Heme is synthesized through a tightly regulated, multistep pathway spanning both the mitochondria and cytosol. Initial and terminal steps of the pathway occur in the mitochondria, whereas intermediate steps take place in the cytosol. In addition to de novo synthesis, cellular heme levels are maintained through intestinal absorption of dietary heme and intercellular transport mechanisms, contributing to systemic heme homeostasis.
Heme functions as a critical prosthetic group in numerous essential proteins. One of the most prominent roles occurs in hemoglobin, where oxygen transport in erythrocytes is facilitated. Additional major hemoproteins include myoglobin (oxygen storage in muscle), cytochromes (electron transport and oxidative phosphorylation), catalase and peroxidases (reactive oxygen species detoxification), and nitric oxide synthase (cell signaling and vascular regulation).
Several structurally distinct forms of heme have been identified, each adapted to specific protein environments. The most abundant form, heme B (protoheme IX), is present in hemoglobin and serves as the precursor for other heme derivatives. Heme A is a modified form present in cytochrome c oxidase (complex IV), whereas heme c is covalently bound to cytochrome c. These specialized heme types are essential for efficient oxidative phosphorylation and ETC function.[3]
The rate-limiting step of heme synthesis is catalyzed by 5′-aminolevulinic acid synthase (ALA synthase, ALAS), which condenses glycine and succinyl-CoA to form δ-aminolevulinic acid (ALA). Pyridoxal phosphate (vitamin B6) functions as a cofactor for this reaction, representing the principal regulatory point of the pathway. Two isoforms of ALAS exist, ALAS1 and ALAS2, each expressed in distinct tissues and regulated by different mechanisms. ALAS1 is a ubiquitous, hepatic mitochondrial isoform regulated primarily by negative feedback inhibition from heme. ALAS2 is an erythroid-specific mitochondrial isoform expressed in developing red blood cells and regulated at the translational level by iron availability, ensuring coordination between heme synthesis and hemoglobin production. The ALAS2 gene is located on the X chromosome, a feature with clinical relevance in X-linked sideroblastic anemia, a disorder characterized by defective heme synthesis secondary to impaired ALAS2 function.[4]
Cellular Level
Porphyrin synthesis is the biochemical process responsible for heme production. The pathway occurs in both the mitochondria and cytosol and consists of 8 enzymatic steps. Heme biosynthesis begins in the mitochondria with the condensation of succinyl-CoA, derived from the citric acid cycle, and glycine to form the key intermediate ALA. This reaction is catalyzed by ALAS, a pyridoxal phosphate–dependent enzyme.[5] This reaction is the rate-limiting and regulatory step of heme synthesis.[6]
ALA molecules formed in the mitochondria are transported into the cytosol, where condensation of 2 ALA molecules produces porphobilinogen (PBG). This reaction is catalyzed by ALA dehydratase, also known as PBG synthase, a zinc-dependent enzyme. The subsequent step involves condensation of 4 PBG molecules to form the linear tetrapyrrole hydroxymethylbilane (HMB), catalyzed by PBG deaminase, also known as HMB synthase.
Cyclization of HMB produces uroporphyrinogen III, a reaction catalyzed by uroporphyrinogen-III synthase. This step is essential, as failure to form the correct isomer results in accumulation of nonphysiologic porphyrins associated with porphyrias. Following synthesis, coproporphyrinogen III is transported back into the mitochondria, where conversion to protoporphyrinogen IX occurs via coproporphyrinogen oxidase. Protoporphyrinogen IX is subsequently oxidized to protoporphyrin IX by protoporphyrinogen oxidase.[7] In the final step, insertion of ferrous iron into protoporphyrin IX is catalyzed by ferrochelatase, resulting in heme formation.[8][9]
Molecular Level
Heme consists of ferrous iron coordinated within protoporphyrin IX, a tetrapyrrole structure composed of 4 pyrrole rings linked by methenyl bridges. In heme B, side chains are arranged around the ring in a specific order—methyl, vinyl, methyl, propionate—with this pattern repeating asymmetrically across the tetrapyrrole structure.
The central ferrous iron exhibits reversible ligand binding, most notably oxygen binding, which underlies the functional role of heme in hemoglobin and myoglobin. Chemical properties of the porphyrin ring, together with the surrounding protein environment, determine ligand affinity and specificity.
At the molecular level, heme exerts an important regulatory function in gene expression and enzyme activity, particularly through feedback inhibition of ALAS (ALAS1) in hepatocytes. Incorporation of iron into protoporphyrin IX by ferrochelatase is essential for heme formation, as disruption of this step (for example, in lead toxicity cases) impairs heme synthesis and results in the accumulation of upstream intermediates.
Function
Heme serves a variety of biological functions as a prosthetic group and cofactor. Heme facilitates oxygen transport in hemoglobin and oxygen storage in myoglobin. The molecule functions as a prosthetic group for CYP450 enzymes and acts as an intracellular reservoir of iron. Within the ETC, heme-containing proteins act as electron shuttles essential for cellular respiration. Heme also participates in signal transduction pathways, including regulation of antioxidant responses, circadian rhythm modulation, and microRNA processing. Heme also plays a role in cellular differentiation and proliferation.
Mechanism
Heme synthesis is differentially regulated in erythroid cells and hepatocytes. This differential regulation reflects distinct functional demands.
In erythroid cells, heme synthesis occurs primarily for incorporation into hemoglobin. Heme promotes globin chain synthesis in developing erythroblasts and reticulocytes, ensuring coordinated production of hemoglobin. Erythropoietin, released by the kidney in response to hypoxia, stimulates erythropoiesis and indirectly enhances heme synthesis. Accumulation of heme within erythroid precursors is required for erythroblast maturation. Regulation in these cells depends largely on intracellular iron availability, which controls the translation of the erythroid-specific enzyme ALAS2. Following terminal differentiation, red blood cells no longer exhibit active heme or hemoglobin synthesis.
In the liver, heme synthesis is more variable and tightly regulated due to the potential toxicity of free heme. Hepatocytes primarily utilize heme for the synthesis of CYP450 enzymes involved in drug metabolism. The hepatic isoform ALAS1 is subject to negative feedback regulation by intracellular heme levels. Decreased intracellular heme concentrations relieve negative feedback on ALAS1, resulting in increased transcription and enzyme activity. Increased demand for CYP450 enzymes, such as that induced by enzyme-inducing drugs, upregulates ALAS1 expression to meet increased heme requirements.
Hemin (oxidized heme) exerts negative feedback on heme synthesis by suppressing ALAS1 through multiple mechanisms. Suppression mechanisms include reduction of ALAS1 gene transcription, destabilization of ALAS1 mRNA, and inhibition of mitochondrial import of ALAS1. These mechanisms ensure tight control of hepatic heme levels and prevent toxic accumulation.
Pathophysiology
Disruptions in the regulation and enzymatic steps of heme synthesis produce a spectrum of clinically significant disorders. Mutation of ALAS2 causes X-linked sideroblastic anemia, characterized by impaired protoporphyrin production and reduced heme synthesis. Iron continues to be transported into developing erythroblasts despite this defect, resulting in mitochondrial iron accumulation and formation of ring sideroblasts.[10]
Abnormalities in downstream enzymes also impair normal porphyrin formation. During heme biosynthesis, the intermediate HMB requires enzymatic conversion to uroporphyrinogen III via uroporphyrinogen III synthase. In the absence of this enzyme, HMB undergoes spontaneous cyclization to form uroporphyrinogen I, which is subsequently converted to coproporphyrinogen I. These compounds are nonphysiologic intermediates and cannot be utilized for heme synthesis, leading to accumulation and contributing to the development of porphyrias.
Clinical Significance
Enzyme deficiencies or impaired substrate availability disrupt heme synthesis, resulting in the accumulation of toxic intermediates in blood, tissues, and urine. This process leads to a group of disorders known as porphyrias (see Image. Enzymatic Steps of Heme Synthesis and Related Porphyrias). These disorders are broadly classified as hepatic or erythropoietic and may present as acute (neurovisceral) or chronic (cutaneous) syndromes. Clinically, porphyrias are characterized by neurovisceral symptoms (eg, abdominal pain, neuropathy, psychiatric disturbances) or photosensitivity, depending on the location of the enzymatic defect. Abnormalities occurring after the formation of HMB lead to the accumulation of porphyrinogens associated with photosensitivity, whereas earlier defects typically present with neurovisceral symptoms.[11] Common features across porphyrias include abdominal pain, tachycardia, hypertension, nausea, confusion, and changes in urine color.
Acute intermittent porphyria results from a deficiency of HMB synthase, leading to the accumulation of ALA and PBG.[12] Clinical presentation includes severe abdominal pain, neuropsychiatric symptoms (eg, irritability, insomnia), tachycardia, and hypertension, without photosensitivity. Urine may darken upon exposure to air due to oxidation of accumulated intermediates.[13] Laboratory findings typically demonstrate elevated urinary PBG levels, with urine darkening upon exposure to air and sunlight. Photosensitivity is absent in acute intermittent porphyria.
Porphyria cutanea tarda, the most common porphyria, results from decreased activity of uroporphyrinogen decarboxylase and may be acquired or inherited. Accumulation of uroporphyrins causes photosensitivity, leading to blistering lesions, hyperpigmentation, and potential hepatic dysfunction. Management includes sunlight avoidance, phlebotomy, and low-dose hydroxychloroquine therapy.
Erythropoietic porphyria (eg, due to ferrochelatase deficiency) is characterized by the accumulation of protoporphyrin IX in erythrocytes. Clinical presentation includes painful photosensitivity with burning, swelling, and itching of sun-exposed skin, with potential progression to hepatic involvement.
Lead poisoning disrupts heme synthesis through the inhibition of ALA dehydratase and ferrochelatase, enzymes dependent on metal cofactors. Inhibition results in the accumulation of ALA and protoporphyrin IX, producing abdominal pain, irritability, fatigue, and neurodevelopmental impairment in children.[14]
Media
(Click Image to Enlarge)
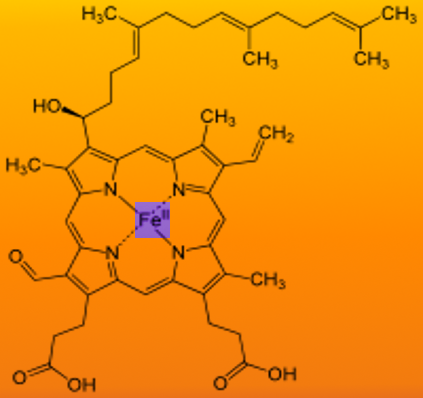
Chemical Structure of Heme A. This diagram illustrates the molecular structure of heme A, a critical component of the cytochrome c oxidase enzyme. The central iron atom is coordinated within a porphyrin ring featuring a long isoprenoid side chain and a formyl group.
Contributed by S Bhimji, MD
(Click Image to Enlarge)
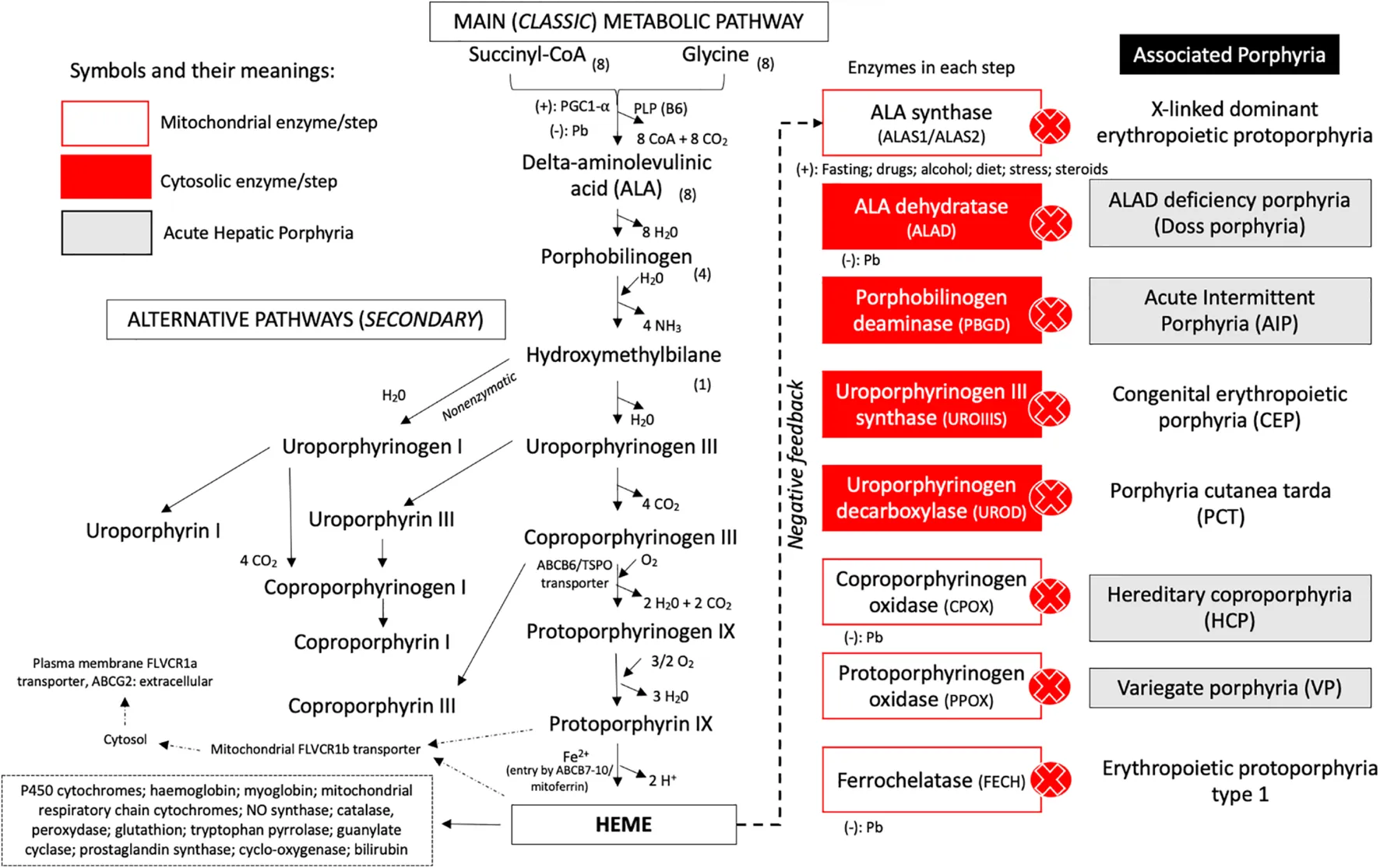
Enzymatic Steps of Heme Synthesis and Related Porphyrias. The diagram illustrates mitochondrial and cytosolic steps of heme biosynthesis, key enzymes at each stage, regulatory features, and corresponding porphyrias linked to enzyme deficiencies.
de Souza PVS, Badia BML, Farias IB, Pinto WBVR, Oliveira ASB. Acute hepatic porphyria: pathophysiological basis of neuromuscular manifestations. Front Neurosci. 2021;15:715523. doi: 10.3389/fnins.2021.715523.
References
Yuan X, Rietzschel N, Kwon H, Walter Nuno AB, Hanna DA, Phillips JD, Raven EL, Reddi AR, Hamza I. Regulation of intracellular heme trafficking revealed by subcellular reporters. Proceedings of the National Academy of Sciences of the United States of America. 2016 Aug 30:113(35):E5144-52. doi: 10.1073/pnas.1609865113. Epub 2016 Aug 15 [PubMed PMID: 27528661]
Shetty T, Corson TW. Mitochondrial Heme Synthesis Enzymes as Therapeutic Targets in Vascular Diseases. Frontiers in pharmacology. 2020:11():1015. doi: 10.3389/fphar.2020.01015. Epub 2020 Jul 15 [PubMed PMID: 32760270]
Belot A, Puy H, Hamza I, Bonkovsky HL. Update on heme biosynthesis, tissue-specific regulation, heme transport, relation to iron metabolism and cellular energy. Liver international : official journal of the International Association for the Study of the Liver. 2024 Sep:44(9):2235-2250. doi: 10.1111/liv.15965. Epub 2024 Jun 18 [PubMed PMID: 38888238]
Medlock AE, Dailey HA. New Avenues of Heme Synthesis Regulation. International journal of molecular sciences. 2022 Jul 5:23(13):. doi: 10.3390/ijms23137467. Epub 2022 Jul 5 [PubMed PMID: 35806474]
Fujiwara T, Harigae H. Biology of Heme in Mammalian Erythroid Cells and Related Disorders. BioMed research international. 2015:2015():278536. doi: 10.1155/2015/278536. Epub 2015 Oct 18 [PubMed PMID: 26557657]
Phillips JD. Heme biosynthesis and the porphyrias. Molecular genetics and metabolism. 2019 Nov:128(3):164-177. doi: 10.1016/j.ymgme.2019.04.008. Epub 2019 Apr 22 [PubMed PMID: 31326287]
Palagyi A, Marais BJ, Abimbola S, Topp SM, McBryde ES, Negin J. Health system preparedness for emerging infectious diseases: A synthesis of the literature. Global public health. 2019 Dec:14(12):1847-1868. doi: 10.1080/17441692.2019.1614645. Epub 2019 May 14 [PubMed PMID: 31084412]
Dunaway LS, Loeb SA, Petrillo S, Tolosano E, Isakson BE. Heme metabolism in nonerythroid cells. The Journal of biological chemistry. 2024 Apr:300(4):107132. doi: 10.1016/j.jbc.2024.107132. Epub 2024 Mar 2 [PubMed PMID: 38432636]
Kim HJ, Khalimonchuk O, Smith PM, Winge DR. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochimica et biophysica acta. 2012 Sep:1823(9):1604-16. doi: 10.1016/j.bbamcr.2012.04.008. Epub 2012 Apr 24 [PubMed PMID: 22554985]
Ono K, Fujiwara T, Saito K, Nishizawa H, Takahashi N, Suzuki C, Ochi T, Kato H, Ishii Y, Onodera K, Ichikawa S, Fukuhara N, Onishi Y, Yokoyama H, Yamada R, Nakamura Y, Igarashi K, Harigae H. Congenital sideroblastic anemia model due to ALAS2 mutation is susceptible to ferroptosis. Scientific reports. 2022 May 30:12(1):9024. doi: 10.1038/s41598-022-12940-9. Epub 2022 May 30 [PubMed PMID: 35637209]
Ricci A, Corradini E, Buzzetti E, Pietrangelo A, Ventura P, Modena Centre of Rare Diseases - Porphyria Working Group. Porphyrias: Pathophysiology and clinical management recommendations for hepatologists. Hepatology communications. 2025 Dec 1:9(12):. doi: 10.1097/HC9.0000000000000822. Epub 2025 Nov 12 [PubMed PMID: 41236019]
Wang B. The acute hepatic porphyrias. Translational gastroenterology and hepatology. 2021:6():24. doi: 10.21037/tgh-2020-01. Epub 2021 Apr 5 [PubMed PMID: 33824928]
Balwani M, Singh P, Seth A, Debnath EM, Naik H, Doheny D, Chen B, Yasuda M, Desnick RJ. Acute Intermittent Porphyria in children: A case report and review of the literature. Molecular genetics and metabolism. 2016 Dec:119(4):295-299. doi: 10.1016/j.ymgme.2016.10.005. Epub 2016 Oct 15 [PubMed PMID: 27769855]
Level 3 (low-level) evidenceSingal AK. Porphyria cutanea tarda: Recent update. Molecular genetics and metabolism. 2019 Nov:128(3):271-281. doi: 10.1016/j.ymgme.2019.01.004. Epub 2019 Jan 18 [PubMed PMID: 30683557]