Introduction
Glucose-6-phosphate dehydrogenase (G6PD) is a cytosolic "housekeeping" enzyme present in all cells that helps prevent damage from reactive oxygen species by generating substrates needed for antioxidative defenses. Erythrocytes are especially vulnerable to reactive oxygen species because they transport oxygen and, once mature, cannot replace damaged proteins. Inherited G6PD deficiency can therefore lead to acute hemolytic anemia during periods of increased oxidative stress.
Triggers include infections, fava beans, and several medications. Antimalarials, in particular, are well known to precipitate hemolysis in affected patients.[1] The Clinical Pharmacogenetics Implementation Consortium has provided a comprehensive, periodically updated list.[2] High- and medium-risk medications that have been associated with hemolytic crises in G6PD deficiency include:
- High-risk medications
- Dapsone
- Methylene blue
- Pegloticase
- Phenazopyridine
- Primaquine (standard dosage)
- Rasburicase
- Tafenoquine
- Toluidine blue
- Medium-risk medications
- Nitrofurantoin
- Primaquine (medium dosage) [2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The G6PD gene, located on the distal long arm of the X chromosome (Xq28), encodes G6PD, and therefore, pathogenic variants follow X-linked inheritance. The gene spans approximately 18 kb, with 13 exons, and produces a 515-amino-acid enzyme.[3] Most disease-causing variants are single-nucleotide missense mutations that result in deficient proteins; small deletions and other variant types are relatively uncommon. Hundreds of variants have been cataloged (well over 200 pathogenic or likely pathogenic, with many additional variants of uncertain significance reported in recent datasets).[1] Complete absence of G6PD activity appears incompatible with life, whereas near-null and severely destabilizing variants present as chronic nonspherocytic hemolytic anemia rather than embryonic demise.[4]
Epidemiology
G6PD deficiency is among the most common human enzymopathies, affecting roughly 400 million people worldwide.[5] Males are affected more often than females because the G6PD gene is X-linked; however, due to lyonization (X-inactivation), heterozygous females can show a wide range of enzyme activity and may be clinically affected.[1] Prevalence is highest across the tropical and subtropical malaria belt—sub-Saharan Africa, the Mediterranean, the Middle East, and South and Southeast Asia—reflecting marked ethnogeographic clustering of variants.[6] Evidence suggests partial, variant- and sex-dependent protection against uncomplicated malaria, whereas protection against severe malaria is uncertain or absent in pooled analyses; the magnitude and direction of effect vary by population.
Pathophysiology
G6PD functions as the catalyst for the rate-limiting first step of the pentose phosphate pathway, using glucose-6-phosphate to convert nicotinamide adenine dinucleotide phosphate (NADP?) into its reduced form, NADPH. See Image. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency. Within red blood cells, NADPH plays an indirect but essential role in protecting cellular structures from injury caused by oxygen-free radicals. Acting as a substrate for glutathione reductase, NADPH supports the production of reduced glutathione, which converts hydrogen peroxide into water. This reaction shields cellular components, particularly the red cell membrane, from oxidative damage.[7]
Reduced G6PD activity prevents red blood cells from producing sufficient NADPH through the pentose phosphate pathway. Inadequate NADPH impairs the ability of glutathione reductase to regenerate reduced glutathione from glutathione disulfide, causing the intracellular glutathione reserve to collapse under oxidative stress. Unneutralized reactive oxygen species then oxidize hemoglobin and membrane structures, leading to the denaturation of hemoglobin into Heinz bodies, which are visible with supravital stains, and to oxidative cross-linking and peroxidation of membrane proteins and lipids. These changes stiffen the membrane and reduce red blood cell deformability.[8]
History and Physical
Most individuals with G6PD deficiency are asymptomatic, but clinical manifestations vary by age and trigger. In newborns, G6PD deficiency is a well-established risk factor for neonatal hyperbilirubinemia; pooled data suggest an estimated 1.6 to 2 times higher risk versus the general population.[9][10] Severe cases disproportionately contribute to kernicterus. In a United States registry analysis, about 20% of infants with kernicterus had G6PD deficiency.[11] Clinically, kernicterus has early, middle, and late stages, and presents with symptoms from lethargy, poor feeding, and hypotonia to irritability, hypertonia, and seizure.
In older children and adults, episodes of hemolysis are triggered by infection, medications, and certain foods, with symptoms including pallor, jaundice, fatigue, dark urine, and even splenomegaly due to red-cell sequestration.[12][13][14]
Evaluation
Newborn Evaluation
Visual inspection alone provides an unreliable assessment of jaundice in newborns; clinicians should therefore obtain an objective bilirubin measurement, eg, total serum bilirubin (TSB), fractionated bilirubin, or transcutaneous bilirubin (TcB). Management should follow hour-specific thresholds outlined in the American Academy of Pediatrics (AAP) hyperbilirubinemia guideline. A TSB measurement should confirm TcB values that approach treatment thresholds, and all treatment decisions should rely on TSB results.
The AAP recommends that G6PD activity testing is indicated in newborns with jaundice of unknown cause, a rise in TSB despite intensive phototherapy, a sudden increase in TSB, a rebound in the TSB level after an initial decline, or when escalation of care becomes necessary. Universal newborn screening for G6PD deficiency is not advised in the United States; however, testing should occur when clinical presentation, ancestry, or family history raises suspicion.[15]
The fluorescent spot test is the most widely used qualitative screening method, but this study may fail to detect heterozygous females. When higher diagnostic precision is required, eg, in females, borderline enzyme activity, or research and policy settings, a quantitative enzyme assay should be performed, either spectrophotometric or through a validated quantitative point-of-care method.[16][17]
Evaluation of Older Children and Adults
Evaluation of older children and adults should begin with a detailed medical history and physical examination, focusing on recent medication use, oxidant exposures, eg, fava beans, and infections. Laboratory findings during oxidant-induced hemolysis typically reveal declining hemoglobin, reticulocytosis, indirect hyperbilirubinemia, elevated lactate dehydrogenase (LDH), and decreased haptoglobin, often with a negative direct antiglobulin test. Peripheral smears may display bite and blister cells, while supravital staining demonstrates Heinz bodies. Confirmation requires a G6PD enzyme activity assay, and molecular testing may be indicated when clarification of the specific variant is needed.[18][19]
Treatment / Management
In neonatal patients, treatment focuses on managing jaundice and preventing kernicterus. This includes phototherapy based on standard published guidelines. In severe cases, an exchange transfusion may be necessary.[15] In older patients, management depends primarily on the overall clinical picture. Less severe presentations may be managed with supportive care and the discontinuation or avoidance of offending agents. Infections should be treated as indicated by history and exam. More severe cases may require transfusions.[20][21](A1)
The optimal care for G6PD deficiency involves an interprofessional approach. Core team members include the pediatrician or internist, hematologist, pharmacist, nursing staff, and the clinical laboratory (for timely enzyme testing and interpretation). Genetics professionals (medical geneticist or genetic counselor) are valuable for variant counseling, carrier testing, especially in females, and family education.
Newborn Management
Priorities include early identification of hyperbilirubinemia, adherence to hour-specific AAP thresholds, prompt phototherapy when indicated, and escalation (eg, NICU transfer, possible exchange transfusion) for rapidly rising or treatment-refractory bilirubin levels. Clinicians should ensure a predischarge bilirubin measurement is performed, arrange time-bound follow-up, and educate caregivers about the warning signs of jaundice in neonates. G6PD activity testing should also be considered in neonatal jaundice that has an early onset, is severe, is poorly responsive to phototherapy, or occurs in infants with suggestive ancestry and family history.
Management in Children and Adults
Management strategies for children and adults with G6PD deficiency depend on the degree of clinical severity.
Mild to moderate episodes
Management focuses on eliminating or avoiding triggers, eg, oxidant medications, fava beans, and naphthalene, while treating concurrent infections. Supportive measures include adequate hydration, rest, and close monitoring of hemoglobin, reticulocyte count, bilirubin, LDH, and haptoglobin levels.
Individuals with severe hemolysis
Patients with significant anemia may require packed red blood cell transfusion. Continuous monitoring for acute kidney injury through urine output and serum creatinine is essential, along with correction of electrolyte and acid–base disturbances. Hospitalization becomes necessary when clinical instability develops.
Special scenarios
Methylene blue must be avoided in suspected or confirmed G6PD deficiency when treating methemoglobinemia, as it can worsen hemolysis. Alternative treatments include high-flow oxygen, intravenous ascorbic acid, transfusion-based therapy, or hyperbaric oxygen in selected cases.
Medication safety and stewardship
Pharmacists, working with prescribers, should review all current and new medications to identify agents with oxidant or hemolytic potential, eg, dapsone, rasburicase, pegloticase, primaquine, and tafenoquine. Caution is warranted with nitrofurantoin and phenazopyridine depending on the clinical context. Patients should receive an updated and accurate list of medications and substances to avoid, and their G6PD status should be clearly documented in the medical record.
Education and prevention
Nurses and clinicians should educate patients and families about recognizing symptoms of hemolysis, eg, dark urine, jaundice, fatigue, and pallor, emphasizing the importance of seeking urgent care when they occur. Counseling should include avoidance of triggers, including specific foods, chemicals, and drugs, and the need to inform all healthcare practitioners of G6PD status before initiating new treatments. Females should be informed about variable expression due to lyonization, which may justify quantitative testing even when qualitative screening appears normal.
Follow-up management
After resolution of an acute episode, clinicians should repeat a quantitative G6PD assay to confirm enzyme deficiency, as testing during hemolysis can yield false-normal results. The patient’s problem list and allergy section should be updated with “G6PD deficiency—risk of hemolysis.” Discharge instructions must include written guidance on avoidance strategies and arrangements for timely follow-up with primary care and hematology clinicians.
Differential Diagnosis
Many disease processes may exhibit similar clinical features of G6PD deficiency. Therefore, differential considerations should include:
- Autoimmune hemolytic anemia
- ABO or Rh hemolytic disease
- Breastmilk Jaundice
- Suboptimal intake hyperbilirubinemia
- Crigler–Najjar syndrome types I and II
- Gilbert syndrome
- Pyruvate kinase deficiency
- Glutathione pathway defects
- Infection-related hemolysis (eg, malaria)
- Hereditary spherocytosis
- Sickle cell anemia
- Thalassemia
Prognosis
Most people with G6PD deficiency have an excellent prognosis. Hemolytic episodes are typically episodic and self-limited when triggers are avoided; chronic hemolysis is uncommon and confined mainly to severe variants. With counseling on oxidant drugs/foods and prompt care during infections, life expectancy is normal.
Complications
In G6PD deficiency, complications cluster around hemolysis. Newborns are prone to significant hyperbilirubinemia with risk of kernicterus. Older patients can develop acute hemolytic anemia after oxidant stress (eg, drugs, infections, fava beans), occasionally with hemoglobinuria and acute kidney injury, and a small subset with severe variants experiences chronic nonspherocytic hemolytic anemia.
Deterrence and Patient Education
G6PD deficiency is an inherited condition that is usually silent until an oxidant trigger precipitates hemolysis. Understanding personal triggers, recognizing warning signs (eg, dark urine, jaundice, or sudden fatigue), and sharing G6PD status with clinicians and pharmacists improves safety across future prescriptions and illnesses. Families of newborns and individuals from higher-prevalence ancestries benefit from awareness of neonatal jaundice risk and from carrying documentation of G6PD status.
Pearls and Other Issues
A special situation exists in the management of G6PD patients with methemoglobinemia. Methemoglobin forms in red blood cells when the iron in the heme group of hemoglobin molecules undergoes oxidation from the normal ferrous (Fe 2+) state to the ferric (Fe3+) state. This ferric state is a poor binder of oxygen. Cyanosis can be seen around 10% methemoglobin. Symptoms, eg, anxiety, light-headedness, and headaches, typically start at a methemoglobin level of 20% or higher, while tachypnea, confusion, and loss of consciousness can be seen with levels between 30% and 50%. Patients with higher levels than those are at risk for seizures, dysrhythmias, metabolic acidosis, and coma. Methemoglobin levels greater than70% are often fatal.[22]
Methemoglobinemia should be considered in patients presenting with central cyanosis and hypoxia whose symptoms are resistant to supplemental oxygen. A specific antidote for severe acute methemoglobinemia is methylene blue. Intravenous methylene blue is reduced to leucomethylene blue through NADPH-dependent mechanisms. Leucomethylene blue is then used as a substrate to reduce methemoglobin back to hemoglobin. However, patients deficient in G6PD lack sufficient NADPH for this mechanism to reduce methylene blue properly. Unreduced methylene blue can cause further oxidative damage in the G6PD-deficient patient, resulting in hemolysis and even death. Therefore, giving methylene blue to patients known or suspected to have G6PD deficiency is contraindicated. Depending on severity, alternative therapies for G6PD-deficient patients presenting with methemoglobinemia include high-flow oxygen, intravenous ascorbic acid, packed red blood cells transfusion, exchange transfusion, or hyperbaric oxygen therapy.[23][24]
Enhancing Healthcare Team Outcomes
G6PD deficiency, an X-linked enzymatic disorder, reduces the ability of red blood cells to withstand oxidative stress. Deficient enzyme activity leads to acute hemolytic anemia during exposure to oxidative triggers such as infections, fava beans, or certain medications, particularly antimalarials and sulfonamides. In newborns, G6PD deficiency increases the risk of severe hyperbilirubinemia and kernicterus, while older patients may experience episodic hemolysis with symptoms including jaundice, fatigue, and dark urine. Early recognition, diagnostic testing, and preventive education remain essential to reducing morbidity and mortality.
Optimal management requires collaboration among physicians, advanced practitioners, nurses, pharmacists, genetic counselors, and laboratory professionals. Clinicians must develop skills in screening, interpreting enzyme assays, managing hemolytic crises, and applying AAP guidelines for neonatal hyperbilirubinemia. Effective interprofessional communication ensures safe medication selection, patient education, and coordinated follow-up. Through teamwork, preventive counseling, and evidence-based care strategies, health professionals can improve patient safety, treatment outcomes, and overall quality of life for those affected by G6PD deficiency.
Media
(Click Image to Enlarge)
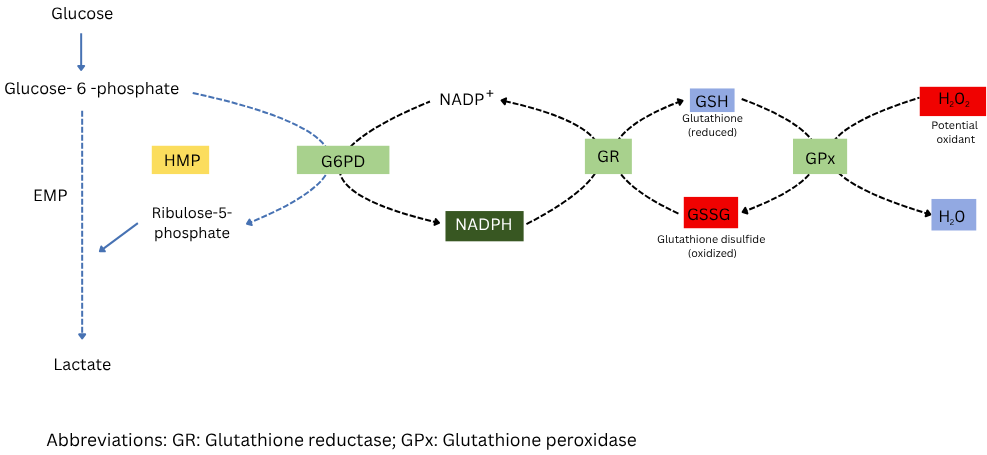
Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency. Diagram showing how G6PD produces NADPH, which helps regenerate reduced glutathione to protect cells from oxidative stress.
Adapted from Shankar V. Pathology of G6PD deficiency. I Love Pathology Web site. https://ilovepathology.com/pathology-of-g6pd-deficiency. Published September 15, 2022. Accessed October 30, 2025.
References
Luzzatto L, Ally M, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Blood. 2020 Sep 10:136(11):1225-1240. doi: 10.1182/blood.2019000944. Epub [PubMed PMID: 32702756]
Gammal RS, Pirmohamed M, Somogyi AA, Morris SA, Formea CM, Elchynski AL, Oshikoya KA, McLeod HL, Haidar CE, Whirl-Carrillo M, Klein TE, Caudle KE, Relling MV. Expanded Clinical Pharmacogenetics Implementation Consortium Guideline for Medication Use in the Context of G6PD Genotype. Clinical pharmacology and therapeutics. 2023 May:113(5):973-985. doi: 10.1002/cpt.2735. Epub 2022 Sep 24 [PubMed PMID: 36049896]
Aung JM, Moon Z, VanBik D, Dinzouna-Boutamba SD, Lee S, Ring Z, Chung DI, Hong Y, Goo YK. Prevalence and molecular analysis of glucose-6-phosphate dehydrogenase deficiency in Chin State, Myanmar. Parasites, hosts and diseases. 2023 May:61(2):154-162. doi: 10.3347/PHD.23004. Epub 2023 May 23 [PubMed PMID: 37258262]
D'Alessandro A, Howie HL, Hay AM, Dziewulska KH, Brown BC, Wither MJ, Karafin M, Stone EF, Spitalnik SL, Hod EA, Francis RO, Fu X, Thomas T, Zimring JC. Hematologic and systemic metabolic alterations due to Mediterranean class II G6PD deficiency in mice. JCI insight. 2021 Jul 22:6(14):. doi: 10.1172/jci.insight.147056. Epub 2021 Jul 22 [PubMed PMID: 34138756]
Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood cells, molecules & diseases. 2009 May-Jun:42(3):267-78. doi: 10.1016/j.bcmd.2008.12.005. Epub 2009 Feb 23 [PubMed PMID: 19233695]
Level 1 (high-level) evidenceHowes RE, Dewi M, Piel FB, Monteiro WM, Battle KE, Messina JP, Sakuntabhai A, Satyagraha AW, Williams TN, Baird JK, Hay SI. Spatial distribution of G6PD deficiency variants across malaria-endemic regions. Malaria journal. 2013 Nov 15:12():418. doi: 10.1186/1475-2875-12-418. Epub 2013 Nov 15 [PubMed PMID: 24228846]
Aziz H, Mohiuddin SS. Biochemistry, Hexose Monophosphate Pathway. StatPearls. 2025 Jan:(): [PubMed PMID: 31869151]
Arese P, Gallo V, Pantaleo A, Turrini F. Life and Death of Glucose-6-Phosphate Dehydrogenase (G6PD) Deficient Erythrocytes - Role of Redox Stress and Band 3 Modifications. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie. 2012 Oct:39(5):328-34. doi: 10.1159/000343123. Epub 2012 Sep 17 [PubMed PMID: 23801924]
Lin Q, Zhu D, Chen C, Feng Y, Shen F, Wu Z. Risk factors for neonatal hyperbilirubinemia: a systematic review and meta-analysis. Translational pediatrics. 2022 Jun:11(6):1001-1009. doi: 10.21037/tp-22-229. Epub [PubMed PMID: 35800274]
Level 1 (high-level) evidenceLiu H, Liu W, Tang X, Wang T. Association between G6PD deficiency and hyperbilirubinemia in neonates: a meta-analysis. Pediatric hematology and oncology. 2015 Mar:32(2):92-8. doi: 10.3109/08880018.2014.887803. Epub 2014 Mar 31 [PubMed PMID: 24684295]
Level 1 (high-level) evidenceCunningham AD, Hwang S, Mochly-Rosen D. Glucose-6-Phosphate Dehydrogenase Deficiency and the Need for a Novel Treatment to Prevent Kernicterus. Clinics in perinatology. 2016 Jun:43(2):341-54. doi: 10.1016/j.clp.2016.01.010. Epub 2016 Feb 28 [PubMed PMID: 27235212]
Frank JE. Diagnosis and management of G6PD deficiency. American family physician. 2005 Oct 1:72(7):1277-82 [PubMed PMID: 16225031]
Almasri G, AlAnazi A, Rahim K, Faqeehi H, Albatati S, Alzabali S. Glucose-6-Phosphate Dehydrogenase Deficiency Presenting as Atypical Hemolytic Uremic Syndrome: A Case Series and Literature Review. Case reports in nephrology. 2025:2025():1938644. doi: 10.1155/crin/1938644. Epub 2025 Sep 25 [PubMed PMID: 41049272]
Level 2 (mid-level) evidenceÖzay HB, Tandoğan M, Dorum BA, Yarcı E. Exchange Transfussion for the Treatment of Severe Indirect Hyperbilirubinemia Caused by Glucose-6-Phosphate Dehydrogenase Deficiency: A Case Report. Fetal and pediatric pathology. 2025 Nov-Dec:44(6):583-588. doi: 10.1080/15513815.2025.2565690. Epub 2025 Sep 27 [PubMed PMID: 41014011]
Level 3 (low-level) evidenceKemper AR, Newman TB, Slaughter JL, Maisels MJ, Watchko JF, Downs SM, Grout RW, Bundy DG, Stark AR, Bogen DL, Holmes AV, Feldman-Winter LB, Bhutani VK, Brown SR, Maradiaga Panayotti GM, Okechukwu K, Rappo PD, Russell TL. Clinical Practice Guideline Revision: Management of Hyperbilirubinemia in the Newborn Infant 35 or More Weeks of Gestation. Pediatrics. 2022 Sep 1:150(3):. pii: e2022058859. doi: 10.1542/peds.2022-058859. Epub [PubMed PMID: 35927462]
Level 1 (high-level) evidenceHalim SA, Bahar R, Abdullah WZ, Zon EM, Yusoff SM. Performance Comparison Between Conventional Fluorescent Spot Test and Quantitative Assay in Detecting G6PD Deficiency in Neonates. Oman medical journal. 2023 Jul:38(4):e524. doi: 10.5001/omj.2023.86. Epub 2023 Jul 31 [PubMed PMID: 37724319]
Pal S, Bansil P, Bancone G, Hrutkay S, Kahn M, Gornsawun G, Penpitchaporn P, Chu CS, Nosten F, Domingo GJ. Evaluation of a Novel Quantitative Test for Glucose-6-Phosphate Dehydrogenase Deficiency: Bringing Quantitative Testing for Glucose-6-Phosphate Dehydrogenase Deficiency Closer to the Patient. The American journal of tropical medicine and hygiene. 2019 Jan:100(1):213-221. doi: 10.4269/ajtmh.18-0612. Epub [PubMed PMID: 30350771]
Phillips J, Henderson AC. Hemolytic Anemia: Evaluation and Differential Diagnosis. American family physician. 2018 Sep 15:98(6):354-361 [PubMed PMID: 30215915]
Herman TF, Killeen RB, Javaid MU. Heinz Body. StatPearls. 2026 Jan:(): [PubMed PMID: 31869086]
Avalos S, Mejia RE, Banegas E, Salinas C, Gutierrez L, Fajardo M, Galo S, Pinto A, Mejia A, Fontecha G. G6PD deficiency, primaquine treatment, and risk of haemolysis in malaria-infected patients. Malaria journal. 2018 Nov 8:17(1):415. doi: 10.1186/s12936-018-2564-2. Epub 2018 Nov 8 [PubMed PMID: 30409136]
Benchimol M, Madeira LB, de Oliveira-Souza R. Late-Life Presentation of Unsuspected G6PD Deficiency. Case reports in critical care. 2018:2018():8198565. doi: 10.1155/2018/8198565. Epub 2018 Sep 25 [PubMed PMID: 30356359]
Level 3 (low-level) evidenceChen RJ, Nappe TM. Methemoglobinemia. StatPearls. 2026 Jan:(): [PubMed PMID: 30726002]
Bistas E, Sanghavi DK. Methylene Blue. StatPearls. 2025 Jan:(): [PubMed PMID: 32491525]
Keats KR, Robinson R, Patel M, Wallace A, Albrecht S. Ascorbic Acid for Methemoglobinemia Treatment: A Case Report and Literature Review. Journal of pharmacy practice. 2024 Aug:37(4):1015-1020. doi: 10.1177/08971900231188834. Epub 2023 Jul 8 [PubMed PMID: 37421600]
Level 3 (low-level) evidence