Introduction
Glomuvenous malformations (GVMs), formerly glomangiomas, are rare cutaneous venous lesions in which clusters of glomus cells (specialized smooth muscle cells) surround dilated venous channels.[1][2] Glomus cells are modified smooth muscle cells derived from the Sucquet–Hoyer canals, which give rise to specialized thermoregulatory structures, the glomus bodies, containing arteriovenous anastomoses.[3] Although glomus bodies are typically located in acral skin, many glomus tumors arise in regions lacking these structures, suggesting that some lesions may originate from pluripotent perivascular or smooth muscle precursor cells.[4] First described by Masson in 1924 and subsequently characterized in detail by Papoff, GVMs represent a distinct subset within the broader spectrum of glomus tumors.[5][6]
Glomuvenous malformations are benign lesions that typically manifest as multiple bluish-purple nodules or plaques. They often are congenital or acquired in early childhood. Unlike solitary glomus tumors, they usually spare the subungual region and show slow progressive enlargement over time.[7][8] Recognizing the clinical behavior and understanding the treatment of these lesions is essential to accurate diagnosis, patient counseling, and long-term management.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Inherited or Familial GVMs
Inherited GVMs follow an autosomal-dominant pattern (~64%) with incomplete penetrance and variable expressivity. Approximately 60% of affected individuals report at least one additional family member with GVMs, with onset typically occurring from birth through adolescence. Penetrance of GLMN gene mutations is estimated at about 80% by age 20 and approaches 100% by age 30.[9][10]
Up to 70% of familial cases are associated with GLMN gene mutations on chromosome 1p21-22, localized to a 4- to 6-cm region identified using yeast artificial chromosome and P1-derived artificial chromosome techniques. At least 14 distinct loss-of-function GLMN mutations have been reported, leading to dysregulated glomus cell proliferation through increased cyclin E and c-Myc expression.[8][11][12][13] The most common pathogenic glomulin mutation, found in 48.8% of inherited cases, is the 157_161del, which causes a frameshift and loss of glomulin function.[14][15] The 4 most frequent mutations display polymorphic markers, suggesting a founder effect for these inherited patterns.[14]
Sporadic GVMs
Sporadic or de novo GVMs (~36%) are not associated with a family history, can also present at birth, and arise from spontaneous genetic mutations.[9] The multifocal and variable nature of GVM reflects the interplay of an inherited germline mutation and an additional somatic mutation that drives lesion development.[1][16]
Epidemiology
Glomuvenous malformations account for approximately 1.6% to 2.0% of all soft tissue tumors and represent nearly 20% of all glomus tumors. Regional GVMs are the most common, while disseminated and plaquelike lesions are exceptionally rare, together accounting for less than 10% of reported cases. Plaque-like GVMs are the rarest, with only about 10 documented cases in the literature.[17] The condition shows no sex prevalence, and about one-third of affected individuals are diagnosed before the age of 20.[11][17][18][19]
Pathophysiology
Glomuvenous malformations are caused by loss-of-function mutations in the GLMN gene on chromosome 1p21-22. Glomulin is critical for vascular morphogenesis, particularly in the differentiation and stabilization of vascular smooth muscle cells.[20] When both GLMN alleles are locally inactivated, glomus cells proliferate around dilated venous channels, producing the characteristic histologic pattern of irregular veinlike vascular spaces surrounded by glomus cells.[1][4][5] Glomuvenous malformations arise from inherited or sporadic mutations and develop when a somatic mutation occurs in addition to a germline GLMN mutation, triggering lesion formation.[1][16] This pattern accounts for the autosomal-dominant inheritance commonly observed and explains why lesions remain localized rather than systemic.
Histopathology
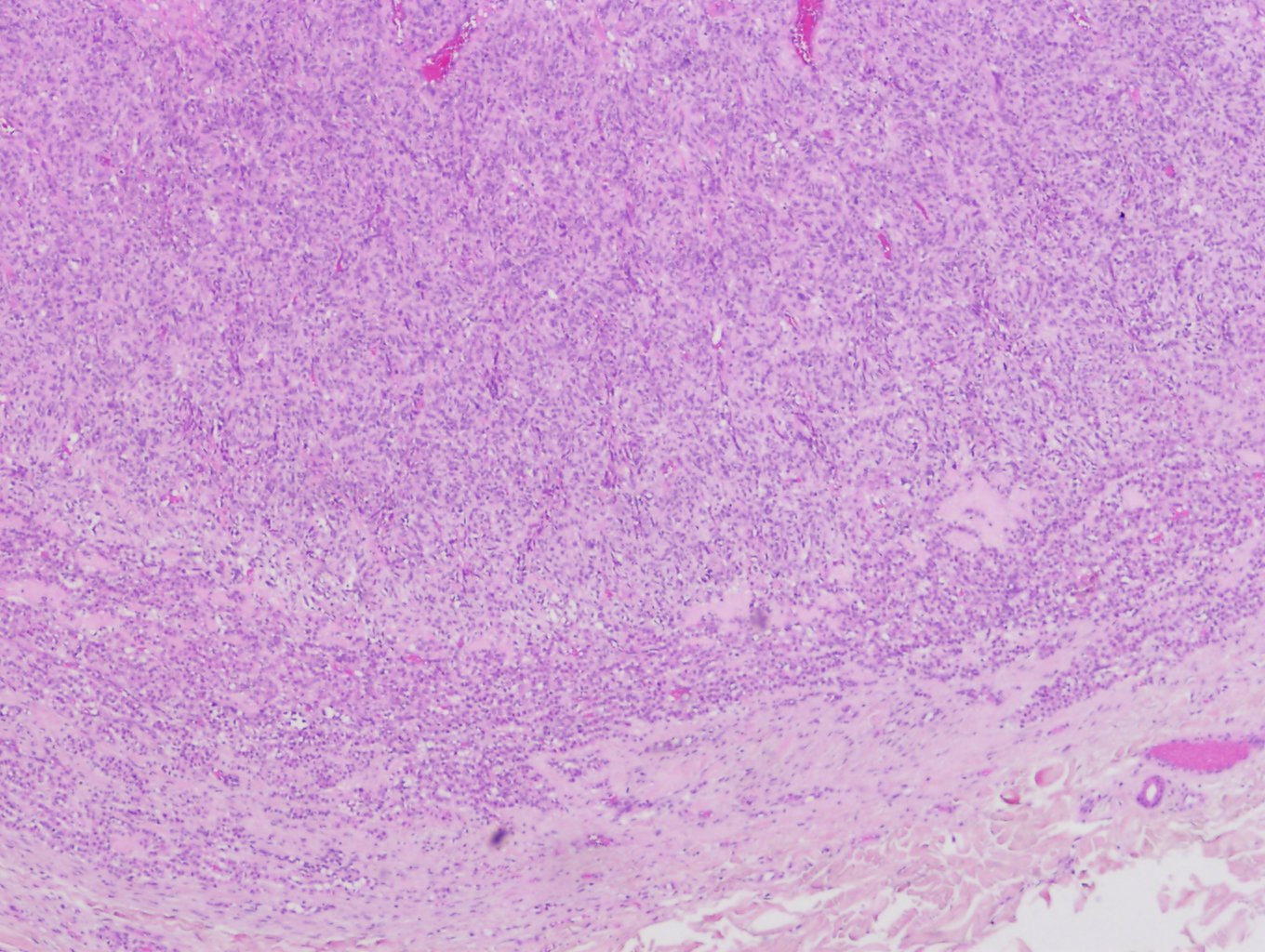
Glomuvenous malformations are characterized histopathologically by irregular dilated vascular channels surrounded by 1 or more layers of glomus cells (see Image. Glomangioma).[9][21] Glomus cells are poorly differentiated smooth muscle–type cells that are positive for vimentin, calponin, and smooth muscle α-actin, and negative for S-100, von Willebrand factor, and desmin.[1][8]
History and Physical
Clinical history can help distinguish GVMs from simple venous malformations. Glomuvenous malformations typically manifest at birth or during early childhood, gradually increasing in size and number over time. Because approximately 63.8% of GVMs are familial and follow an autosomal-dominant inheritance pattern, obtaining a thorough family history during evaluation is crucial.
Glomuvenous malformations are characteristically painful on compression or palpation, which can help distinguish them from simple venous malformations, in which pain typically occurs upon awakening, after physical activity, or in response to hormonal fluctuations. Glomuvenous malformations tend not to worsen during puberty, menstrual cycles, or pregnancy. Notably, elastic compression garments generally worsen pain in patients with GVMs but typically provide relief to those with simple venous malformations (see Table 1).[9][22]
Table 1. Glomuvenous Malformations vs Venous Malformations
|
|
Glomuvenous Malformations |
Simple Venous Malformations |
|
Family History |
Often familial (63.8%) |
Rare (1.2%) |
|
Inheritance Pattern |
Autosomal dominant with incomplete penetrance |
Usually sporadic, rarely autosomal dominant (TIE2 mutation) |
|
Age of Onset |
Present at birth or childhood, may appear before age 30 |
Present at birth, may enlarge over time |
|
Appearance |
Nodular and scattered, or plaquelike and segmental; raised and cobblestonelike with minor hyperkeratosis |
Soft, blue, often localized vascular lesions |
|
Distribution |
Predominantly cutaneous, commonly involves extremities, may extend into subcutis (50% have subfascial extension) |
Involves skin, subcutis, and mucosa; may affect muscle, bone, joints, and internal organs |
|
Mucosal Involvement |
Usually not found in the mucosa |
Common (mucosal or subcutaneous lesions) |
|
Compressibility |
Incompletely compressible |
Readily compressible, decreases with external pressure and limb elevation |
|
Pain trigger |
Painful on compression or palpation |
Painful on awakening, after activity, or with hormonal changes |
|
Pain Response to Compression |
Pain worsens |
Pain improves |
|
Hormonal Influence |
None |
More than 50% cases have increased pain with puberty, menstrual cycles, anticontraceptive drugs, or pregnancy |
Glomuvenous malformations can be classified into 2 main subtypes: multiple nodular GVMs and plaquelike (segmental) GVMs. The multiple nodular form is more common, accounting for 90% of reported cases. Glomuvenous malformations typically present in childhood as bluish-purple lesions arranged in a cobblestonelike pattern.
The lesions are often papular or nodular, hyperkeratotic, and range from 2 to 10 mm in diameter, though their size and number can vary considerably. Tenderness on palpation is common, with pain frequently triggered by external pressure or cold exposure.[1][9] Lesions predominantly manifest in areas that are rich in glomus bodies, especially the distal extremities, such as the palms, wrists, forearms, feet, and subungual regions. Approximately 75% of cases involve the hands.[11]
Glomuvenous malformations have traditionally been considered confined to the skin; however, a retrospective study found that approximately 50% of patients demonstrate subfascial extension. The most common site of involvement is intramuscular, followed by intraosseous and, less frequently, intra-articular locations.[22] Visceral involvement is extremely rare but has been reported in the nasal cavity; mediastinum; gastrointestinal, respiratory, and urogenital tracts; and hepatobiliary system.
Additionally, some patients with GVMs have presented with congenital cardiac anomalies such as ventricular septal defects and transposition of the great arteries.[7][23][24] Although glomus bodies are absent in normal nerve tissue, rare cases of glomangioma involving nerves have been reported.[25] Tracheal lesions, though rare, may cause dyspnea, hemoptysis, and retrosternal chest pain.[26]
Plaquelike GVMs are rare, accounting for approximately 10% of recorded cases. They manifest as confluent, pink, plaquelike lesions that appear in infancy. The lesions rapidly worsen, turning thick and darkening to purple or dark blue.[9] The differences between the subtypes are summarized in Table 2 below.
Table 2. Comparison of Multiple Nodular and Plaquelike (Segmental) GVM Subtypes
|
|
Multiple Nodular Global Vascular Management |
Plaquelike (Segmental) Global Vascular Management |
|
Appearance |
Scattered bluish papules and nodules |
Confluent plaquelike lesions, pink to purplish-blue |
|
Age of Onset |
Childhood, grows slowly with age |
Present at birth or infancy, rapidly worsens |
|
Location |
Predominantly the extremities |
Can be extensive, segmental distribution |
|
Texture |
Raised, cobblestonelike, hyperkeratotic |
Thickened, plaquelike |
Evaluation
All patients with clinical features of GVMs should undergo imaging evaluation. Ultrasonography with duplex Doppler is typically the initial modality, as it can differentiate low-flow from high-flow malformations and distinguish solid from cystic components. MRI with and without intravenous contrast is then used to assess the full extent of deep or diffuse lesions and to further characterize complex anatomy.[27][28] Dynamic, time-resolved, contrast-enhanced magnetic resonance angiography can further characterize lesion vascularity.
This modality demonstrates features such as subtle arterial enhancement, early venous shunting, and progressive filling of dilated venous spaces, which help distinguish GVMs from simple venous malformations.[27] Imaging can support but does not confirm the diagnosis of GVM. Definitive diagnosis requires histopathology or genetic testing. When surgical excision or biopsy is performed, histopathologic examination typically demonstrates irregular vascular spaces lined by glomus cells.[9][21] Immunohistochemical staining for smooth muscle markers can support the diagnosis.[1][8]
Genetic testing is recommended for patients with a personal or family history of GVM. This typically involves analysis of the GLMN gene on chromosome 1p21–22 to identify both germline and somatic mutations.[20] Four common glomulin mutations are responsible for about 70% of familial cases, with the 157delAAGAA mutation found in nearly half of affected families.[14] Identification of these mutations confirms the diagnosis, guides management, limits unnecessary testing, and enables genetic counseling. Families should be informed that penetrance increases from approximately 80% by age 20 to nearly 100% by age 30.[10]
Treatment / Management
Treatment options include observation, surgical excision, laser therapy, sclerotherapy, embolization, or combination therapy. Treatment should be individualized based on lesion characteristics (location, size, depth), the presence of pain or other symptoms, patient age, proximity to vital structures, and patient concerns, including cosmetic or functional impact. For asymptomatic lesions, monitoring and observation are recommended. Surgery is indicated for lesions causing persistent pain or cosmetic disfigurement and well-defined lesions that spare vital structures.[29] Embolization can serve as a preoperative adjunct to facilitate surgical resection or in cases where considerable intraoperative hemorrhage is anticipated.[30]
Sclerotherapy may also be used as a surgical adjunct, similarly to embolization. This modality was trialed as a standalone treatment; however, it is less effective for large GVMs than for typical VMs.[31] Facial GVMs, in particular, respond less effectively to sclerotherapy than simple venous malformations. This diminished response is thought to result from the presence of glomus cells, which may limit the effectiveness of sclerosing agents.[9][29](B2)
Laser treatment has emerged as an effective option for GVMs (see Table 3), particularly for superficial lesions located on acral sites and the trunk.[32] The Alexandrite laser (755 nm) appears to be the most effective overall, producing optimal outcomes in clinical flattening, clearance of blue-red vascular coloration, and pain resolution, while also causing the least procedural and postprocedural discomfort.[31] The Nd:YAG laser (1064 nm) is particularly effective for palmar GVMs and has been successfully used for mucosal lesions; the long-pulsed Nd:YAG laser offers a safe and effective alternative, especially for GVMs in adolescents.[31][33]
Additionally, a dual pulsed-dye laser–Nd:YAG approach has demonstrated at least a 60% reduction in lesion size in most patients, with good tolerability and rare adverse effects.[31] The greatest clinical response to laser treatment is observed for GVMs located on acral sites and the trunk, with sustained clearance following therapy. For extensive or complex lesions, combination approaches may be considered, including preoperative sclerotherapy or embolization followed by surgical excision, or a combination of laser and surgical techniques.
Table 3. Treatment Options for Global Vascular Management
|
Treatment Modality |
Indications |
Technique/Agents |
Efficacy |
Advantages |
Limitations |
|
Surgical excision |
Symptomatic, localized lesions; persistent pain; cosmetic disfigurement; well-defined lesions sparing vital structures |
Complete resection of the lesion; may use preoperative embolization with N-butyl cyanoacrylate to minimize blood loss |
Curative for localized lesions; no recurrence after complete excision |
Definitive treatment; eliminates the lesion completely |
Intraoperative hemorrhage risk; limited by proximity to vital structures; not suitable for extensive lesions |
|
Alexandrite (755-nm) laser |
Superficial global vascular management (GVMs), acral sites, and trunk lesions |
Employ settings to minimize scarring risk |
Most effective overall for clinical flattening; clearance of blue-red coloration, and pain resolution |
Lowest procedural and postprocedural pain, sustained clearance; effective for acral and trunk lesions |
Limited to superficial lesions |
|
Nd:YAG (1064-nm) laser |
Superficial and mucosal GVMs, palmar lesions |
Long-pulsed photothermolysis |
Effective for palmar GVMs and mucosal lesions; ameliorates discoloration and lesion size |
Safe and effective, especially in adolescents; can treat mucosal lesions |
May require multiple sessions |
|
Dual pulsed dye laser–Nd:YAG laser |
Superficial GVMs |
Combined approach |
≥60% reduction in lesion size in the majority of patients |
Good tolerability, rare adverse effects, can be combined with sclerotherapy |
Limited to superficial lesions |
|
Sclerotherapy |
Large or extensive GVMs (limited effectiveness compared to simple venous malformations) |
Sodium tetradecyl sulfate, polidocanol, ethanol, bleomycin |
Disappointing results in large GVMs, facial GVMs respond less effectively than simple venous malformations |
Minimally invasive, can be repeated |
Less effective than in simple venous malformations due to the presence of glomus cells; risk of extravasation and complications |
|
Preoperative sclerotherapy + surgery |
Complex or extensive lesions, lesions near vital structures |
Sclerotherapy followed by surgical excision after fibrosis develops (several months) |
Facilitates easier resection, improved outcomes for complex lesions |
Sclerotherapy-induced fibrosis makes subsequent resection easier, reduces blood loss |
Requires staged procedures, longer treatment timeline |
|
Nd:YAG laser + sclerotherapy |
Superficial and deeper components |
Combined Nd:YAG laser and sclerotherapy at the same anesthetic setting |
Addresses both superficial discoloration and deeper vascular components |
Single anesthetic session possible; comprehensive treatment |
Requires careful coordination; airway protection is paramount due to posttreatment swelling |
|
Embolization + surgery |
Localized GVMs with considerable vascularity |
Preoperative embolization with N-butyl cyanoacrylate immediately before surgical resection |
Facilitates complete resection while minimizing blood loss |
Single-stage procedure possible, reduced intraoperative hemorrhage |
Requires specialized expertise, limited availability |
Differential Diagnosis
The differential diagnosis for GVMs includes several vascular anomalies and tumors with similar bluish, nodular, or plaquelike skin lesions. Simple venous malformations are soft, blue, compressible lesions that shrink with elevation or pressure. They often involve muscles and joints, causing pain upon awakening or with hormonal changes, whereas GVMs are painful to the touch and primarily affect the skin and subcutaneous tissue of the extremities.[9]
Blue rubber bleb nevus syndrome features multiple venous malformations and hemangiomas of the skin and internal organs, particularly the gastrointestinal tract. Lesions are soft, compressible, and blue; patients often experience gastrointestinal bleeding and iron-deficiency anemia, whereas cutaneous lesions are typically asymptomatic.[34] Solitary glomus tumors are benign vascular masses, usually subungual, causing pain, tenderness, and cold sensitivity. Unlike GVMs, these tumors are neither familial nor multiple and may be misdiagnosed as hemangiomas on imaging.[35]
Tufted angiomas are rare vascular tumors presenting as dusky red or blue plaques or nodules with a characteristic doughnut appearance, typically arising in infancy on the thorax, neck, or shoulders. They enlarge slowly and rarely regress, with about one-third being tender.[36] Pyogenic granulomas are rapidly growing, friable papules or nodules (yellow, red, or purple) that frequently bleed or ulcerate. They commonly arise on the head and neck in children or on the trunk and extremities in adults and often regress spontaneously, distinguishing them from GVMs.[37]
Syndromic associations include Maffucci syndrome and Cobb syndrome. Maffucci syndrome features multiple enchondromas with hemangiomas or simple venous malformations, skeletal deformities, limb shortening, and fractures.[38] Cobb syndrome is characterized by cutaneous vascular lesions on the torso with segmental spinal vascular malformations and neurological symptoms such as pain, weakness, or numbness.[39]
Key diagnoses not to miss include Kaposi sarcoma, angiosarcoma, and epithelioid hemangioendothelioma. Kaposi sarcoma is a multicentric tumor with purplish lesions on the skin, mucosa, gastrointestinal tract, lymph nodes, lungs, and bones, confirmed by biopsy and immunohistochemistry for human herpesvirus 8, particularly in patients with HIV, transplant recipients, or those from endemic regions.[40] Angiosarcoma is a malignant vascular tumor characterized by infiltrative growth of abnormal endothelial cells, diagnosed by biopsy using endothelial markers (von Willebrand factor, CD31, CD34) and the absence of melanocytic markers.[41] Epithelioid hemangioendothelioma is a rare vascular tumor characterized by spindle cells and a specific genetic marker (WWTR1-CAMTA1 fusion), often presenting as multiple pulmonary nodules, confirmed by biopsy and molecular testing for gene fusions.[42]
Prognosis
The prognosis for GVMs is generally favorable, as they are benign vascular malformations without malignant potential.[20] Morbidity from GVMs arises primarily from pain on compression, cosmetic disfigurement, and anatomic distortion when lesions are large or involve critical structures. Complete excision is associated with low recurrence rates and excellent symptom resolution.[6][20] Long-term outcomes depend on early diagnosis, complete surgical removal, and close follow-up to monitor for recurrence or complications.
Complications
Glomuvenous malformations may lead to clinical complications, including pain, cosmetic disfigurement, anatomic distortion, and progressive growth. If untreated, GVMs typically continue to cause pain and increase in size and number as the patient grows. Cosmetic concerns are most common with lesions affecting the face and extremities.
In plaquelike GVM, a case of chylous ascites has been reported; however, visceral involvement remains extremely rare.[17] Recurrence following surgical excision occurs in approximately 10% to 33% of cases, often due to incomplete removal or multifocality of the lesions.[12][18] Lesions may require multiple treatment approaches to achieve optimal results.
Deterrence and Patient Education
Patients should be advised to monitor for any signs of lesion recurrence, increased size, or new symptoms. Prompt follow-up with a healthcare professional is essential if such changes occur, as they may indicate progression or complications requiring further evaluation and possible intervention.
Pearls and Other Issues
Diagnosing GVMs can be challenging due to their clinical overlap with other vascular anomalies and soft tissue tumors, such as simple venous malformations, hemangiomas, blue rubber bleb nevus syndrome, and other cutaneous lesions. Misdiagnosis may delay appropriate treatment and lead to unnecessary interventions. A thorough patient history, including family history, and a detailed physical examination are critical. Imaging studies and, when appropriate, histopathologic analysis help confirm the diagnosis.
Enhancing Healthcare Team Outcomes
Glomuvenous malformations often resemble other dermatologic lesions, such as papules or nodules, making accurate diagnosis and management a multidisciplinary effort. Clinicians and advanced practitioners play a central role in evaluating clinical features, interpreting imaging and histopathology, developing evidence-based treatment plans, and coordinating referrals to appropriate specialists. Radiologists contribute by assessing lesion depth, vascular involvement, and atypical features on imaging studies, guiding therapeutic decision-making.
Specialty care nurses facilitate patient-centered care by coordinating follow-up, monitoring postprocedural outcomes, and educating patients and families on symptom recognition, recurrence, and self-care. Genetic counselors provide guidance in cases with suspected familial patterns. Effective interprofessional collaboration, ethical shared decision-making, and clear communication among the care team enhance diagnostic accuracy, optimize treatment outcomes, ensure patient safety, and improve overall satisfaction.
Media
(Click Image to Enlarge)
Glomangioma Contributed by Daniel Neelon, MD
References
Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibé M, Warman ML, Tan OT, Olsen BR, Vikkula M. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations ("glomangiomas"). American journal of human genetics. 2002 Apr:70(4):866-74 [PubMed PMID: 11845407]
McMahon MH, Tahir N, Balasubramanian M. GLMN causing vascular malformations: the clinical and genetic differentiation of cutaneous venous malformations. BMJ case reports. 2022 Jun 22:15(6):. doi: 10.1136/bcr-2021-246114. Epub 2022 Jun 22 [PubMed PMID: 35732373]
Level 3 (low-level) evidenceTaaffe A, Barker D, Wyatt EH, Bury HP. Glomus tumours: a clinico-pathological survey. Clinical and experimental dermatology. 1980 Jun:5(2):219-25 [PubMed PMID: 6254702]
Level 3 (low-level) evidenceRequena L, Sangueza OP. Cutaneous vascular proliferation. Part II. Hyperplasias and benign neoplasms. Journal of the American Academy of Dermatology. 1997 Dec:37(6):887-919; quiz 920-2 [PubMed PMID: 9418757]
Brauer JA, Anolik R, Tzu J, Meehan S, Lieber CD, Geronemus RG. Glomuvenous Malformations (Familial generalized multiple glomangiomas). Dermatology online journal. 2011 Oct 15:17(10):9 [PubMed PMID: 22031635]
Arens C, Dreyer T, Eistert B, Glanz H. Glomangioma of the nasal cavity. Case report and literature review. ORL; journal for oto-rhino-laryngology and its related specialties. 1997 May-Jun:59(3):179-81 [PubMed PMID: 9186975]
Level 3 (low-level) evidenceTewattanarat N, Srinakarin J, Wongwiwatchai J, Areemit S, Komvilaisak P, Ungarreevittaya P, Intarawichian P. Imaging of a glomus tumor of the liver in a child. Radiology case reports. 2020 Apr:15(4):311-315. doi: 10.1016/j.radcr.2019.12.014. Epub 2020 Jan 20 [PubMed PMID: 31988680]
Level 3 (low-level) evidenceLeger M, Patel U, Mandal R, Walters R, Cook K, Haimovic A, Franks AG Jr. Glomangioma. Dermatology online journal. 2010 Nov 15:16(11):11 [PubMed PMID: 21163162]
Level 3 (low-level) evidenceBoon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Archives of dermatology. 2004 Aug:140(8):971-6 [PubMed PMID: 15313813]
Level 2 (mid-level) evidenceBorroni RG, Grassi S, Diegoli M, Grasso M, Arbustini E. Incomplete penetrance of GLMN gene c.395-1G}C mutation in a family with glomuvenous malformations. International journal of dermatology. 2014 Nov:53(11):1362-4. doi: 10.1111/ijd.12588. Epub 2014 Jun 25 [PubMed PMID: 24961656]
Souza NGA, Nai GA, Wedy GF, Abreu MAMM. Congenital plaque-like glomangioma: report of two cases. Anais brasileiros de dermatologia. 2017:92(5 Suppl 1):43-46. doi: 10.1590/abd1806-4841.20175766. Epub [PubMed PMID: 29267443]
Level 3 (low-level) evidenceCabral CR, Oliveira Filho Jd, Matsumoto JL, Cignachi S, Tebet AC, Nasser Kda R. Type 2 segmental glomangioma--Case report. Anais brasileiros de dermatologia. 2015 May-Jun:90(3 Suppl 1):97-100. doi: 10.1590/abd1806-4841.20152483. Epub [PubMed PMID: 26312686]
Level 3 (low-level) evidenceJha A, Khunger N, Malarvizhi K, Ramesh V, Singh A. Familial Disseminated Cutaneous Glomuvenous Malformation: Treatment with Polidocanol Sclerotherapy. Journal of cutaneous and aesthetic surgery. 2016 Oct-Dec:9(4):266-269. doi: 10.4103/0974-2077.197083. Epub [PubMed PMID: 28163461]
Brouillard P, Ghassibé M, Penington A, Boon LM, Dompmartin A, Temple IK, Cordisco M, Adams D, Piette F, Harper JI, Syed S, Boralevi F, Taïeb A, Danda S, Baselga E, Enjolras O, Mulliken JB, Vikkula M. Four common glomulin mutations cause two thirds of glomuvenous malformations ("familial glomangiomas"): evidence for a founder effect. Journal of medical genetics. 2005 Feb:42(2):e13 [PubMed PMID: 15689436]
Suárez-Magdalena O, Monteagudo B, Figueroa O, Gómez-Pérez MI. Glomulin gene c.157_161del mutation in a family with multiple glomuvenous malformations. International journal of dermatology. 2019 Feb:58(2):e43-e45. doi: 10.1111/ijd.14312. Epub 2018 Nov 21 [PubMed PMID: 30460983]
Amyere M, Aerts V, Brouillard P, McIntyre BA, Duhoux FP, Wassef M, Enjolras O, Mulliken JB, Devuyst O, Antoine-Poirel H, Boon LM, Vikkula M. Somatic uniparental isodisomy explains multifocality of glomuvenous malformations. American journal of human genetics. 2013 Feb 7:92(2):188-96. doi: 10.1016/j.ajhg.2012.12.017. Epub 2013 Jan 31 [PubMed PMID: 23375657]
Mallory SB, Enjolras O, Boon LM, Rogers E, Berk DR, Blei F, Baselga E, Ros AM, Vikkula M. Congenital plaque-type glomuvenous malformations presenting in childhood. Archives of dermatology. 2006 Jul:142(7):892-6 [PubMed PMID: 16847206]
Gonçalves R, Lopes A, Júlio C, Durão C, de Mello RA. Knee glomangioma: a rare location for a glomus tumor. Rare tumors. 2014 Oct 27:6(4):5588. doi: 10.4081/rt.2014.5588. Epub 2014 Dec 18 [PubMed PMID: 25568752]
Level 3 (low-level) evidenceTony G, Hauxwell S, Nair N, Harrison DA, Richards PJ. Large plaque-like glomangioma in a patient with multiple glomus tumours: review of imaging and histology. Clinical and experimental dermatology. 2013 Oct:38(7):693-700. doi: 10.1111/ced.12122. Epub [PubMed PMID: 24073652]
Level 3 (low-level) evidenceIsch EL, Guler M, Self DM, Caterson EJ. Symptomatic Glomuvenous Malformation of the Anterior Chest: Clinical Presentation, Surgical Management, and Genetic Considerations. The Journal of craniofacial surgery. 2025 May 15:():. doi: 10.1097/SCS.0000000000011504. Epub 2025 May 15 [PubMed PMID: 40372364]
Borroni RG, Grassi S, Concardi M, Puccio I, Giordano C, Agozzino M, Caspani C, Grasso M, Diegoli M, Arbustini E. Glomuvenous malformations with smooth muscle and eccrine glands: unusual histopathologic features in a familial setting. Journal of cutaneous pathology. 2014 Mar:41(3):308-15. doi: 10.1111/cup.12283. Epub 2014 Jan 20 [PubMed PMID: 24345188]
Shaikh R, Alomari AI, Mulliken JB, Fishman SJ, Kozakewich HP, Chaudry G. Subfascial involvement in glomuvenous malformation. Skeletal radiology. 2014 Jul:43(7):895-7. doi: 10.1007/s00256-014-1836-3. Epub 2014 Feb 28 [PubMed PMID: 24577796]
Cullen RD, Hanna EY. Intranasal glomangioma. American journal of otolaryngology. 2000 Nov-Dec:21(6):402-4 [PubMed PMID: 11115526]
Level 3 (low-level) evidenceGoujon E, Cordoro KM, Barat M, Rousseau T, Brouillard P, Vikkula M, Frieden IJ, Vabres P. Congenital plaque-type glomuvenous malformations associated with fetal pleural effusion and ascites. Pediatric dermatology. 2011 Sep-Oct:28(5):528-31. doi: 10.1111/j.1525-1470.2010.01216.x. Epub 2010 Dec 7 [PubMed PMID: 21133993]
Level 3 (low-level) evidenceScheithauer BW, Rodriguez FJ, Spinner RJ, Dyck PJ, Salem A, Edelman FL, Amrami KK, Fu YS. Glomus tumor and glomangioma of the nerve. Report of two cases. Journal of neurosurgery. 2008 Feb:108(2):348-56. doi: 10.3171/JNS/2008/108/2/0348. Epub [PubMed PMID: 18240933]
Level 3 (low-level) evidenceParker KL, Zervos MD, Donington JS, Shukla PS, Bizekis CS. Tracheal glomangioma in a patient with asthma and chest pain. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010 Jan 10:28(2):e9-e10. doi: 10.1200/JCO.2009.22.7942. Epub 2009 Oct 26 [PubMed PMID: 19858390]
Level 3 (low-level) evidenceFlors L, Norton PT, Hagspiel KD. Glomuvenous malformation: magnetic resonance imaging findings. Pediatric radiology. 2015 Feb:45(2):286-90. doi: 10.1007/s00247-014-3086-x. Epub 2014 Jul 5 [PubMed PMID: 24996811]
Level 3 (low-level) evidenceExpert Panel on Pediatric Imaging, Bardo DME, Gill AE, Iyer RS, Chan SS, Cooper ML, Dasgupta RA, Guimaraes CV, Hammer MR, Krowchuk DP, Levin TL, Liang MG, Meyers ML, Samet JD, Sammer MBK, Schooler GR, Squires JH, Sura AS, Trout AT, Pruthi S. ACR Appropriateness Criteria® Soft Tissue Vascular Anomalies: Vascular Malformations and Infantile Vascular Tumors (Non-CNS)-Child. Journal of the American College of Radiology : JACR. 2024 Jun:21(6S):S310-S325. doi: 10.1016/j.jacr.2024.02.030. Epub [PubMed PMID: 38823953]
Gallant SC, Chewning RH, Orbach DB, Trenor CC 3rd, Cunningham MJ. Contemporary Management of Vascular Anomalies of the Head and Neck-Part 1: Vascular Malformations: A Review. JAMA otolaryngology-- head & neck surgery. 2021 Feb 1:147(2):197-206. doi: 10.1001/jamaoto.2020.4353. Epub [PubMed PMID: 33237296]
Tieu DD, Ghodke BV, Vo NJ, Perkins JA. Single-stage excision of localized head and neck venous malformations using preoperative glue embolization. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2013 Apr:148(4):678-84. doi: 10.1177/0194599813475586. Epub 2013 Jan 28 [PubMed PMID: 23358955]
Moreno-Arrones OM, Jimenez N, Alegre-Sanchez A, Fonda P, Boixeda P. Glomuvenous malformations: dual PDL-Nd:YAG laser approach. Lasers in medical science. 2018 Dec:33(9):2007-2010. doi: 10.1007/s10103-018-2493-x. Epub 2018 Mar 29 [PubMed PMID: 29594737]
Fisher C, Gadarowski MB, Roberts J, Hivnor C. Successful Multi-Modal Laser Intervention and Histopathological Evaluation of Multiple Glomangiomas. Lasers in surgery and medicine. 2025 Jan:57(1):41-45. doi: 10.1002/lsm.23867. Epub 2024 Dec 29 [PubMed PMID: 39737544]
Trost J, Buckley C, Smidt AC. Long-Pulsed Neodymium-Doped Yttrium Aluminum Garnet Laser for Glomuvenous Malformations in Adolescents. Pediatric dermatology. 2015 Sep-Oct:32(5):e217-8. doi: 10.1111/pde.12631. Epub 2015 Jul 2 [PubMed PMID: 26138991]
Jin XL, Wang ZH, Xiao XB, Huang LS, Zhao XY. Blue rubber bleb nevus syndrome: a case report and literature review. World journal of gastroenterology. 2014 Dec 7:20(45):17254-9. doi: 10.3748/wjg.v20.i45.17254. Epub [PubMed PMID: 25493043]
Level 3 (low-level) evidenceKunz EM, Ram B. Atypical Presentation of a Glomus Tumor in the Rearfoot: A Case Report. Journal of the American Podiatric Medical Association. 2020 Nov 1:110(6):. pii: Article_15. doi: 10.7547/19-184. Epub [PubMed PMID: 33301580]
Level 3 (low-level) evidenceMetry DW, Hebert AA. Benign cutaneous vascular tumors of infancy: when to worry, what to do. Archives of dermatology. 2000 Jul:136(7):905-14 [PubMed PMID: 10890993]
Shen-Wagner J, Amidon J, Carek S. Diagnosing Common Benign Skin Tumors. American family physician. 2024 Oct:110(4):353-361 [PubMed PMID: 39418568]
Prokopchuk O, Andres S, Becker K, Holzapfel K, Hartmann D, Friess H. Maffucci syndrome and neoplasms: a case report and review of the literature. BMC research notes. 2016 Feb 27:9():126. doi: 10.1186/s13104-016-1913-x. Epub 2016 Feb 27 [PubMed PMID: 26920730]
Level 3 (low-level) evidencePutilina M, Teplova N, Dvornikov A. Cutaneomeningospinal Angiomatosis (Cobb Syndrome) in a Young Patient. CNS & neurological disorders drug targets. 2021:20(10):888-893. doi: 10.2174/1871527320666210218083550. Epub [PubMed PMID: 33602108]
Yarchoan R, Uldrick TS. HIV-Associated Cancers and Related Diseases. The New England journal of medicine. 2018 Mar 15:378(11):1029-1041. doi: 10.1056/NEJMra1615896. Epub [PubMed PMID: 29539283]
Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. The Lancet. Oncology. 2010 Oct:11(10):983-91. doi: 10.1016/S1470-2045(10)70023-1. Epub 2010 May 25 [PubMed PMID: 20537949]
Tang B, Zhao S, Wang J, Xiang M, Dai J, Liu Q. Pulmonary epithelioid haemangioendothelioma: A comprehensive review of clinical and molecular advances. Clinics (Sao Paulo, Brazil). 2025 Jan-Dec:80():100789. doi: 10.1016/j.clinsp.2025.100789. Epub 2025 Oct 13 [PubMed PMID: 41086575]
Level 3 (low-level) evidence