Introduction
Fahr disease is named after Karl Theodor Fahr, a German neurologist who first reported the disorder in 1930.[1] This rare neurological condition is characterized by abnormal idiopathic calcification of the basal ganglia, the most commonly affected site, and is most often inherited in an autosomal dominant pattern. Abnormal calcified deposits, composed of calcium carbonate and phosphate, are not just limited to the basal ganglia but also occur in some other locations, such as the thalamus, hippocampus, dentate nucleus, cerebral cortex, and cerebellar subcortical white matter.
Although the terms Fahr disease and Fahr syndrome are often used interchangeably in the literature, some authors distinguish between the 2 as follows:
- For the familial or genetic form (previously considered idiopathic), the term Fahr disease should be used. This condition is also known as bilateral strio-pallido-dentate calcinosis, primary familial brain calcification, or calcinosis nucleorum.
- For secondary causes of basal ganglia calcifications with known underlying causes, such as metabolic, endocrine, or systemic disorders, the term Fahr syndrome should be used.[2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Fahr disease is most commonly inherited in an autosomal dominant pattern with incomplete and age-related penetrance, but it may also be transmitted as an autosomal recessive trait or occur sporadically. Some studies have reported genetic anticipation in this disease.[3] Fahr disease, also known as primary familial brain calcification, most often has an autosomal dominant inheritance pattern. Solute carrier family 20 (phosphate transporter) member 2 (SLC20A2), platelet-derived growth factor subunit B (PDGFB), platelet-derived growth factor subunit receptor B (PDGFRB), and xenotropic and polytropic retrovirus receptor 1 (XPR1) genes are implicated.[4][5][6] Approximately 46% of cases have unknown gene mutations. Other loci linked to Fahr disease include the IBGC1 locus on chromosome 14q, a locus on chromosome 2q, and another on chromosome 8.[1][7] Autosomal recessive inheritance involves the myogenesis-regulating genes glycosidase (MYORG), junctional adhesion molecule 2 (JAM2), and N-α-acetyltransferase 60 (NAA60).[5] The adult phenotype involves more than 50 heterozygous mutations in the solute carrier family 20 member 2 gene (SLC20A2).[1][4] Conversely, the pediatric phenotype involves homozygous mutations.[8]
The pathogenesis of Fahr syndrome primarily involves disarray in calcium-phosphate homeostasis.[4] The common etiologies include:
Epidemiology
The true prevalence of Fahr disease is largely unknown because of insufficient investigation of patients' first-degree relatives. Fahr disease has a prevalence of less than 1 in 1,000,000 people, with a male to female ratio of 2:1.[1] Fahr disease is reported to commonly affect people in their 40s and 50s. Patients are typically in good health in their youth and tend to develop this progressive neurodegenerative disease later in adulthood.
Pathophysiology
Abnormal calcium deposition in Fahr syndrome is hypothesized to result from either abnormal brain calcium metabolism or metastatic deposition because of locally altered blood-brain barrier permeability. Defective iron transport and free radical production cause tissue damage, which initiates calcification around a nidus composed of mucopolysaccharides and related substances. Calcium deposition starts within the vessel wall and perivascular space, and then slowly extends to involve the entire neuron. Progressive calcification compresses nearby vessels, reducing blood flow and consequently continuing the vicious cycle of decreased blood flow, tissue injury, and mineral deposition.[7] High levels of copper, zinc, magnesium, and iron, as well as altered glucose metabolism in the basal ganglia, have also been reported. Additionally, increased cerebrospinal fluid levels of the central nervous system (CNS)-specific peptide homocarnosine and low histidine levels have been observed in some cases.
Pathogenesis in Fahr disease
The SLC20A2 gene encodes the transmembrane type III sodium-dependent phosphate transporter 2 (PiT2), which is predominantly expressed in regions of the globus pallidus, thalamus, and cerebellum. The mutation thereby leads to the deposition of inorganic phosphate in the extracellular matrix alongside calcium.[8] The resultant alteration in the calcium-phosphate ratio leads to pericyte injury and the precipitation of calcium hydroxyapatite crystals within the tunica media of small- and medium-sized cerebral arteries and capillaries.[5][10]
Histopathology
On gross pathologic examination, the affected areas show granular deposits and solid nodules, accompanied by mild cortical atrophy. On histological examination, concentrically deposited calcium along the walls of small- and medium-sized arteries is observed, with less common involvement of veins. Sometimes, droplet calcifications can be observed. Ischemic changes and diffuse gliosis can be observed surrounding large deposits. On electron microscopy, calcium appears as amorphous or crystalline material surrounded by a basal membrane. Calcium granules are visible in the cytoplasm of neuronal and glial cells.
History and Physical
Approximately 25% of patients with Fahr disease remain asymptomatic.[6] Incidental detections occur in 0.3% to 1.2% of brain computed tomography (CT) scans.[10] Most adult patients become symptomatic in their fourth or fifth decade of life.[10] The main patterns of presentation include neuropsychiatric symptoms and cognitive decline.[6] Motor dysfunctions, including parkinsonian-like, choreic, and cerebellar, are also common.[1][11] Patients may present acutely with symptoms similar to schizophrenia, mania, or medical emergencies such as seizures, encephalopathy, and strokes.[4][12] Lethargy, low mood, irritability, tingling sensations, frequent muscle twitches, and cramps may occur because of hypoparathyroidism. Clinical examination may reveal positive results for the Chvostek and Trousseau tests.[13] Chronic renal disease and nephrocalcinosis are also common.[4][13] Mutations in KIF1A, FZD8, and PDGFA may be associated with dental abnormalities.[14] Pediatric cohorts may present with delayed milestones, growth retardation, and convulsions, and may have bilateral cataracts and microcephaly.[8] The salient clinical features in the disease process include:
Movement Disorder–Like Features
- Signs and symptoms resembling parkinsonism, such as bradykinesia, rigidity, tremor, hypophonia, hypomimia, mask-like facies, and shuffling gait
- Clumsiness
- Fatigability
- Gait dysfunction
- Choreoathetosis
- Dystonia
- Slurred speech
- Muscle cramping
- Pyramidal or cerebellar symptoms (rarely) [15]
Neuropsychiatric Features
- Depression
- Apoplexia
- Dementia, mostly frontal-executive type resembling subcortical dementia in Wilson disease and Huntington disease
- Concentration deficits
- Behavioral changes [16]
Other Neurologic Features
- Loss of consciousness
- Tetany
- Seizures
- Spasticity
- Speech impairment
- Myoclonus
- Coma
- Papilledema
- Chronic headache
- Vertigo
- Urinary urgency or incontinence
- Impotence
- Severe hypertension [7][17]
In a study by Batla et al involving 137 patients, a clinical correlation with genomic abnormalities (genotype-phenotype relationship) was established. Patients with SLC20A2 mutations were more likely to have parkinsonism and involvement of the thalamus and dentate nucleus. Patients with PDGFB mutations were observed to have more frequent headaches and white matter cysts.[18]
Physical Examination
Physical examination should include:
- General physical examination
- Complete neurological examination
- Mental health screening
- Memory and cognitive assessment
- Functional outcome measures
- Functional Independence Measure
- Dynamic Gait Index
- Timed Up and Go Test
- Fullerton Advanced Balance Scale
Evaluation
The clinical diagnostic criteria for Fahr disease are as follows:
- Bilateral calcification of the basal ganglia on neuroimaging
- Progressive symptomatology, most often starting in the fourth or fifth decade of life
- Absence of metabolic abnormalities, endocrinopathies, or mitochondrial disease
- Exclusion of other mimics, such as infections, toxins, or trauma
- Positive family history [8][19][20]
To meet the diagnostic criteria, the following investigations are typically performed to exclude other causes of brain calcifications.
Imaging Studies
Bilateral basal ganglia calcification is visualized in both Fahr disease and secondary causes.
- Brain CT scan: A brain CT scan is the most sensitive modality for localizing and assessing the extent of calcium deposits. These deposits are most commonly observed within the basal ganglia, the gray-white interface, the cerebellum, and the thalamus.[13] The total calcification visual rating score may help assess the severity of calcification on CT (see Image. Fahr Disease).[21]
- MRI brain: Magnetic resonance imaging (MRI) provides better anatomic details but is less sensitive than a CT scan. Calcified deposits show low-intensity signals on T2-weighted images and high- to low-intensity signals on T1-weighted images. However, a study by Kozic et al found heterogeneous signal intensities in some subjects, which can lead to misinterpretations.
- Fluorodeoxyglucose positron emission tomography: May show decreased fluorodeoxyglucose F18 uptake, particularly in the basal ganglia.
- Plain skull radiograph: Calcifications can be visualized. However, this modality is less sensitive than a CT scan.
Laboratory Investigations
Routine laboratory results are within the reference range in patients with Fahr disease. Any abnormality should prompt further investigation to rule out secondary causes of brain calcifications. Laboratory investigations include:
- Routine hemogram and complete metabolic panel
- Blood and urine heavy metal levels
- Serum calcium, phosphorus, magnesium, alkaline phosphatase, calcitonin, vitamin D, and parathyroid hormone (PTH) levels
- Cerebrospinal fluid analysis for bacteria, viruses, and parasites
- Ellsworth-Howard test demonstrates a 10- to 20-fold increase in urinary cyclic adenosine monophosphate levels following a 200-U PTH injection
- Autoimmune panel studies including antineutrophil cytoplasmic antibodies, antinuclear antibodies, and complement C3 levels
- Pyruvate and lactate levels for mitochondrial disease and TORCH screening in pediatric patients [4][8]
Molecular Genetic Testing
Clinicians perform this testing in an index case that meets the diagnostic criteria to establish the diagnosis of Fahr disease.[4][8] The 3 approaches to genomic testing include:
- Serial gene testing: Sequentially testing for genes one at a time, based on the frequency of causation (SLC20A2>PDGFB>PDGFRB>XPR1)
- Multigene panel: One-time analysis of all 4 implicated pathogenic variants (SLC20A2, PDGFB, PDGFRB, XPR1) and other genes to rule out secondary syndromes
- Comprehensive genomic testing
Treatment / Management
To date, no definitive cure is available for Fahr disease, as is the case with other neurodegenerative disorders, and treatment is focused primarily on symptomatic relief. Treatment is mostly patient-centered and symptomatic, with an aim of metabolic correction.[22][23] Interdisciplinary management is essential.[24] Daily calcium and vitamin D supplementation remains the mainstays of management.[13] Bisphosphonate therapy has shown promise.[8] Gene therapy may provide newer insights into the management of PFBC gene mutation.[25] Continuum surveillance, genetic counseling, and family screening are of paramount importance in Fahr disease.[26] Structured follow-up and adherence counseling are pivotal.[4](B3)
The management aspects of the disease should include:
Symptomatic Management
- Antiepileptic therapy for seizures
- Painkillers for headaches
- Anticholinergics for urinary urgency or incontinence
- Selective serotonin reuptake inhibitors for depression, obsessive-compulsive behaviors, and anxiety
- Neuroleptics for movement disorders
Cautionary Measures
The use of carbamazepine, benzodiazepines, and barbiturates in patients with Fahr disease can lead to increased gait dysfunction. Psychiatrists and neurologists should exercise great caution with the use of antidepressants and anxiolytics because the threshold for adverse effects with these drugs is low in patients with Fahr disease. Lithium has been known to increase seizure risk in these patients, and neuroleptics can exacerbate extrapyramidal symptoms.
Physical Rehabilitation
- Maintaining range of motion and preventing contractures with range-of-motion exercises, passive stretching, and facilitated stretching can improve mobility.
- Strengthening underused muscles: Antigravity extensor muscles are often at risk of weakening in neurological disease. A general conditioning program and the use of exercise machines may be helpful.
- Improving postural stability: This intervention involves correcting sitting postures. Patients with basal ganglia dysfunction can perform balance training while engaging in activities such as using stairs, reaching out, and similar tasks, and progress from wide to narrow bases, from static to dynamic, or from low to higher levels of cognition.
- Gait dysfunction: Assess for assistive devices, auditory cues (eg, counting), or visual cues (eg, a laser attached to canes).
- Symptomatic improvement: Relaxation techniques for anxiety, deep brain stimulation for hyperkinetic disorders, soft tissue release for spasticity, or sensory stimulation in basal ganglia dysfunction.[27]
Genetic Counseling
Fahr disease is transmitted in an autosomal dominant pattern. However, the family history is not fully conclusive. Sometimes the family history of a patient is unrevealing because of the early death of a parent before the disease became evident, or the parent may have had late-onset disease or reduced penetrance.
No demonstrated medical benefits exist for screening relatives. However, predictive testing may be offered to asymptomatic first-degree relatives older than 18 after careful consideration of the ethical and psychosocial implications, particularly because no curative treatment is available. Testing options include a brain CT scan, in which calcium deposits precede clinical manifestations by years, and molecular testing when a PDGFB, PDGFRB, XPR1, or SLC20A2 pathogenic variant has been identified in the proband. Testing individuals older than 18 years can help them make decisions about marriage, family planning, career, and finances. Testing is typically not recommended in individuals younger than 18, considering social issues, discrimination, and anxiety in the future, negative impact on the parent-child or sibling-child relationship, and uncertain CT scan predictability, as penetrance is age-dependent.
Prenatal Counseling
Prenatal testing and preimplantation genetic diagnosis are possible for a high-risk pregnancy in which a PDGFB, PDGFRB, XPR1, or SLC20A2 pathogenic variant has been detected in the proband.
Surveillance
Annual neurologic and neuropsychiatric assessment should be performed.
Differential Diagnosis
Basal ganglia calcifications occur in several other familial and nonfamilial conditions that must be excluded before diagnosing primary familial basal ganglia calcification. These conditions include:
Endocrine Disorders
- Parathyroid hormone disturbances: PTH disturbances are the most common cause of bilateral basal ganglia calcifications. Reduced PTH levels cause hyperphosphatemia and hypocalcemia, which promote calcification. In a study involving 150 patients with Fahr syndrome, 35 (23.3%) had idiopathic hypoparathyroidism and 23 (15.3%) had secondary (postthyroidectomy) hypoparathyroidism.[28][29]
- Hypoparathyroidism: This condition may be idiopathic, resulting from congenital absence, fatty replacement, or atrophy of the parathyroid glands, or secondary, most commonly due to inadvertent removal during thyroidectomy.
- Pseudohypoparathyroidism and pseudopseudohypoparathyroidism: Both conditions are phenotypic variants with autosomal dominant inheritance and mutation of the GNAS gene. Both have clinical features of Albright hereditary osteodystrophy. In pseudohypoparathyroidism, resistance to the action of parathyroid hormone results in hypocalcemia, hyperphosphatemia, and elevated PTH levels, whereas pseudopseudohypoparathyroidism has no biochemical abnormalities, with normal calcium and phosphorus levels and a normal response to PTH secretion.
- Kenny-Caffey syndrome type 1: This rare skeletal disorder is caused by a mutation in the TBCE gene and is characterized by dwarfism, cortical thickening and medullary stenosis of the long bones, facial dysmorphism, hypocalcemia because of congenital hypoparathyroidism, and basal ganglia calcifications.[30]
Mitochondrial Myopathy
Basal ganglia calcification is a recognized finding in mitochondriopathy due to abnormal calcium homeostasis.[31] In a study, neuroradiological findings were examined in 6 families comprising 24 individuals with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS); bilateral basal ganglia calcification was the most common radiological finding.
Infectious Diseases
- Intrauterine and perinatal infections: Herpes, cytomegalovirus, rubella, and toxoplasmosis may cause basal ganglia and intracerebral calcification in newborns, often presenting with microcephaly, seizures, chorioretinitis, or other CNS manifestations.
- Brucellosis: A study by Mousa et al showed that 9 (13.8%) of 65 patients with CNS brucellosis (neuropsychiatric manifestations) had radiologic evidence of basal ganglia calcifications. Calcifications were unilateral in 3 patients, bilaterally symmetric in 4, and bilaterally asymmetric in 2.
- Toxoplasmosis: The basal ganglia are affected in approximately 75% of patients, typically showing multiple ring-enhancing lesions with edema and occasional calcifications.
- Neurocysticercosis: Larval cysts may sometimes occur in the basal ganglia and may become calcified.
- HIV/AIDS: In a neuropathology study involving 52 patients with HIV, researchers found that 3 patients had basal ganglia calcifications.
Congenital Disorders
- Cockayne syndrome or Neill-Dingwall syndrome: This autosomal recessive disorder is characterized by an underlying defect in DNA repair.[32] Cockayne syndrome type 1 is the classic variant, in which initially normal development occurs, followed by features that emerge after the first 2 years of life, including hearing and visual impairment, CNS and peripheral nervous system degeneration, and bilateral basal ganglia calcifications. Cockayne syndrome type 2 is a more severe congenital variant, also known as cerebro-oculofacial syndrome or Pena-Shokeir type 2 syndrome. No postnatal neurological development occurs, with associated eye and skeletal anomalies.
- Aicardi-Goutières syndrome: This neurodevelopmental disorder is characterized by microcephaly, neonatal seizures, cerebral atrophy, encephalopathy, and cerebral calcifications, typically bilateral and in the basal ganglia, especially the putamen and globus pallidus.
- Tuberous sclerosis complex: This autosomal dominant neurocutaneous disorder is caused by mutations in the TSC1 and TSC2 genes. Clinical manifestations involve multiple organ systems, including the skin, with ash-leaf macules, shagreen patches, facial angiofibromas, and adenoma sebaceum; the kidneys, with angiomyolipomas; the heart, with rhabdomyosarcoma; and the central nervous system, with subependymal nodules, hamartomas, and cortical and subcortical tubers, which may calcify. The frequency of calcifications increases with age.
- Coats plus syndrome (cerebroretinal microangiopathy with calcifications and cysts): Coats disease is a congenital nonhereditary eye disorder characterized by abnormal development of blood vessels behind the retina, resulting in complete or partial blindness. Coats plus disease has associated neurologic features with intracerebral calcifications and formation of parenchymal cysts, bone, and gastrointestinal abnormalities.
- Down syndrome: In a study by Takashima et al, among 33 patients with Down syndrome, 45% had basal ganglia calcifications, most commonly in the globus pallidus, and this proportion increased with age.[33]
- Other conditions: Other conditions in the differential diagnosis include immunodeficiency, in which 38 patients have been reported to have basal ganglia calcification, and pantothenate kinase-associated neurodegeneration.
Adult-Onset Disorders
- Neuroferritinopathy: This autosomal dominant disease is a variant of neurodegeneration with brain iron accumulation, presenting with adult-onset dystonia and cognitive and behavioral changes, with radiographic evidence of excessive iron storage and cystic degeneration of the putamen.
- Spinocerebellar ataxia type 20: This autosomal dominant disease shows marked calcifications in the cerebellum, particularly the dentate nucleus.
- Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (Nasu-Hakola disease): This autosomal recessive disease presents with fractures, dementia, and bilateral basal ganglia calcifications, particularly the putamen.
- Other conditions: Other considerations include diffuse neurofibrillary tangles with calcification (Kosaka-Shibayama disease) and dentatorubral-pallidoluysian atrophy (Haw River syndrome).[34]
Dermatological Conditions
- Lipoid proteinosis: This rare genodermatosis involves intracellular accumulation of amorphous hyaline material in the skin, mucous membranes, and internal organs. Manifestations include alopecia, photosensitivity, dwarfism, seizures, and dementia. Selective brain parenchymal calcifications occur in the amygdala, corpus striatum, hippocampus, and parahippocampal gyrus.[35]
- Dyskeratosis congenita: This disorder presents with a classic triad of dysplastic nails, lacy reticular pigmentation of the upper chest, and oral leukoplakia. Intracranial calcifications have been reported in the Revesz syndrome variant.[36]
Toxins
Neural necrosis has been reported with excess vitamin D, lead, mercury, ionizing radiation, and methotrexate therapy.
Normal Aging
Approximately 0.3% to 1.5% of brain CT scans incidentally show basal ganglia calcifications. In a study by Forstl et al, about 70% of autopsies showed microscopic calcifications in the globus pallidus and the dentate nucleus, yet patients were typically asymptomatic.[37] In a study by Ostling et al, basal ganglia calcification was strongly correlated with psychotic symptoms in older adults.[38]
Other Conditions
Other reported conditions associated with brain calcifications include biotinidase deficiency, hereditary folate malabsorption, carbonic anhydrase deficiency, celiac disease, cerebral lupus, and tetrahydrobiopterin-deficient hyperphenylalaninemia.[39]
The most common mimics of the entity can be summarized as follows:
- Physiologic: Aging
- Metabolic: Hypoparathyroidism, pseudohypoparathyroidism, chronic hypocalcemia, and vitamin D deficiency
- Genetic: Aicardi-Goutières syndrome, Cockayne syndrome, Krabbe disease, Sturge-Weber syndrome, tuberous sclerosis, and MELAS syndrome
- Infections: Cytomegalovirus infection, toxoplasmosis, Zika virus infection, Cryptococcus neoformans infection, neurocysticercosis
- Tumor: Oligodendroglioma, craniopharyngioma, germ cell tumors, central neurocytoma, and chondroma
- Autoimmune: Systemic lupus erythematosus
- Inflammatory: Sarcoidosis
- Intoxications: Carbon monoxide, manganese toxicity, and lead poisoning
- Postradiation
- Neurodegenerative conditions: Parkinsonism
- Idiopathic [5][9][11][13][40]
Prognosis
The prognosis of a patient with Fahr disease is variable and unpredictable. No correlation exists between the age at disease onset, symptoms at onset, or extent of brain calcifications and disease severity.[1] Penetrance is age-dependent, reaching approximately 95% by 50 years of age. Therefore, an asymptomatic disease carrier may present with a negative CT scan finding at a younger age. Calcifications have been reported to become more extensive with age, as evidenced by follow-up scans of confirmed cases.
Complications
Although Fahr disease is a rare disorder, it has some crippling complications. Clinicians should be aware of the following complications:
- Movement disorders
- Neuropsychiatric issues such as depression, dementia, and psychosis
- Speech difficulty
- Encephalopathy
- Epilepsy
- Strokes
- Recurrent falls and orthopedic consequences, including contractures
Consultations
The following are some advisable consultations to ensure the best outcomes for patients with Fahr disease:
- Psychiatrist
- Neurologist
- Speech therapist
- Urologist
- Dietician
- Otolaryngologist
- Physical therapist
- Orthopedics
Deterrence and Patient Education
A diagnosis of a chronic and progressive neurological disease can be a frightening experience for many patients. Educating patients is crucial for the long-term management and follow-up of Fahr disease due to its incurable and progressive nature.
Support Groups
A patient may experience a wide range of emotions, such as fear, anger, depression, and anxiety, after the diagnosis. Talking to other people with other neurodegenerative diseases may help them to share experiences and information.
Physical and Exercise Therapy
Exercise can help patients feel better physically and mentally. This activity helps improve balance, flexibility, and strength; prevent contractures; and enhance quality of life and socialization. Strengthening and stretching exercises should be advised. Studies have shown that aerobic exercises such as walking, riding a stationary bicycle, and water aerobics are energizing for patients with parkinsonian features.
Fall Prevention
Patients and caregivers are encouraged to make the home as safe as possible by:
- Eliminating loose rugs, which lead to tripping
- Adequate lighting
- Using grab bars in showers and bathtubs
- Decreasing clutter in the house
Driving Safety
If no motor symptoms are present, patients can drive, but driving ability should be reevaluated periodically, especially if motor or cognitive decline is observed. If patients start developing seizures, they are encouraged to discontinue driving. Clinicians should advise patients to use other means of transportation, such as walking, cabs, public buses, and trains.
Pearls and Other Issues
Fahr disease can present in autosomal dominant, familial, or sporadic forms. A recent genetic study identified mutations in SLC20A2—a gene in the IBGC3 region that encodes type III sodium-dependent phosphate transporter 2 (PiT2)—as a primary cause of Fahr disease, which must be distinguished from Fahr syndrome, in which basal ganglia calcification is secondary. Pathologic basal ganglia calcification can be caused by idiopathic hypoparathyroidism, secondary hypoparathyroidism, hyperparathyroidism, postthyroidectomy status, birth anoxia, cysticercosis, toxoplasmosis, calcified infarction, HIV infection, and other factors. In general, endocrine, toxic, metabolic, or degenerative etiologies cause widespread and symmetrical intracranial calcification. Conversely, pathologic calcified lesions caused by infectious illness, vascular injuries, or neoplasms are often dispersed and asymmetrical in size and location.
No specific therapy for Fahr disease has been shown to slow the progression of brain calcification. Treatment is often symptomatic. A trial of a CNS-specific calcium channel blocker, such as nimodipine, failed to demonstrate effectiveness.[14] In some preclinical investigations, disodium etidronate, a bisphosphonate, provided functional benefit and symptomatic relief without reducing the number of calcifications. Substantial calcium deposits in affected arteries may lead to small artery blockage and, ultimately, ischemic stroke or transient ischemic attack.[41]
Enhancing Healthcare Team Outcomes
Because Fahr disease is a rare, chronic, progressive disorder with no cure, coordinated interprofessional management is essential to optimize patient outcomes. The care team typically includes clinicians, nurses, pharmacists, social workers, and physical therapists. Treatment focuses on symptom control, preservation of functional independence, and improvement of quality of life.
Nurses play a central role in patient and caregiver education, symptom monitoring, fall-risk assessment, and care coordination. Nurses are often the first to recognize functional decline or new neurologic symptoms and can facilitate timely referral to appropriate specialists. Pharmacists help optimize medication therapy by identifying potential drug interactions, minimizing adverse effects, and addressing the challenges of polypharmacy. Rehabilitation therapists develop individualized programs to improve mobility, balance, communication, and activities of daily living. Social workers provide psychosocial support, connect patients and families with community resources and long-term care services, and assist with advance care planning, financial counseling, and disability benefits when appropriate.
Media
(Click Image to Enlarge)
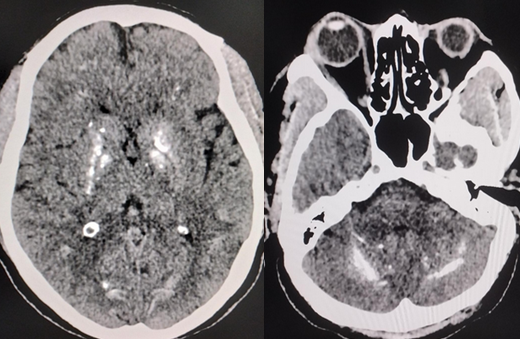
Fahr Disease. Axial noncontrast CT images of the brain demonstrate bilateral, symmetric calcifications involving the basal ganglia and dentate nuclei, characteristic findings of Fahr disease.
Contributed by S Munakomi, MD
References
Nisar S, Mushtaq H, Khan MH, Sajjad F, Afridi A, Bacha Z, Hafza B, Kamil KA. Fahr's Syndrome in a Young Adult Male: A Case of Seizure and Cognitive Decline Secondary to Hypoparathyroidism. Clinical case reports. 2026 Mar:14(3):e72206. doi: 10.1002/ccr3.72206. Epub 2026 Mar 11 [PubMed PMID: 42016627]
Level 3 (low-level) evidencePerugula ML, Lippmann S. Fahr's Disease or Fahr's Syndrome? Innovations in clinical neuroscience. 2016 Jul-Aug:13(7-8):45-6 [PubMed PMID: 27672489]
Maeda K, Idehara R, Nakamura H, Hirai A. Anticipation of familial idiopathic basal ganglia calcification? Internal medicine (Tokyo, Japan). 2012:51(8):987 [PubMed PMID: 22504267]
Level 3 (low-level) evidenceAlrashidi FS, Mohajab AH. Basal Ganglia Calcification Secondary to Postsurgical Hypoparathyroidism Presenting With Seizures and Acute Confusion: A Case Report (Fahr Syndrome). Cureus. 2026 Jan:18(1):e100553. doi: 10.7759/cureus.100553. Epub 2026 Jan 1 [PubMed PMID: 41625059]
Level 3 (low-level) evidencePosa A, Schob S, Wohlgemuth WA, Kornhuber ME. Bilateral Cerebral Calcifications in Secondary Fahr's Syndrome. Case reports in neurological medicine. 2025:2025():5598992. doi: 10.1155/crnm/5598992. Epub 2025 Dec 20 [PubMed PMID: 41473459]
Level 3 (low-level) evidenceYang D, Lu Y, Huang H, Chen Y, Jiang Z, Yao R, Bu Y, Li Y, Cen Z, Luo W. Gene Variants Related to Primary Familial Brain Calcification: Perspectives from Bibliometrics and Meta-Analysis. eNeuro. 2025 Jun:12(6):. pii: ENEURO.0058-25.2025. doi: 10.1523/ENEURO.0058-25.2025. Epub 2025 Jun 23 [PubMed PMID: 40456615]
Level 1 (high-level) evidenceGeschwind DH, Loginov M, Stern JM. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification (Fahr disease). American journal of human genetics. 1999 Sep:65(3):764-72 [PubMed PMID: 10441584]
Sufrani MA. First case of Fahr's disease with homozygous mutations of SLC20A2, among the Libyan children. BMC pediatrics. 2026 Jan 24:26(1):. doi: 10.1186/s12887-026-06524-z. Epub 2026 Jan 24 [PubMed PMID: 41580645]
Level 3 (low-level) evidenceVallejo Ruiz M, Moral Presa I. [A case report of severe hypocalcemia secondary to Fahr's syndrome]. Anales del sistema sanitario de Navarra. 2026 Feb 18:49(1):. pii: e1138. doi: 10.23938/ASSN.1138. Epub 2026 Feb 18 [PubMed PMID: 41704128]
Level 3 (low-level) evidenceNimodia D, Parihar PH, Dudhe S, Patil R, Bhangale PN, Kotla R. Navigating diagnostic uncertainty in fahr's disease: a case report with neuroimaging correlations. Radiology case reports. 2025 Feb:20(2):1252-1256. doi: 10.1016/j.radcr.2024.11.016. Epub 2024 Dec 6 [PubMed PMID: 39717218]
Level 3 (low-level) evidenceBoudriot ET, Hainc N, Balint B. Basal Ganglia and Prominent Cortical Contouring Calcification in SLC20A2-Related Primary Familial Brain Calcification. Movement disorders clinical practice. 2025 Feb:12(2):258-259. doi: 10.1002/mdc3.14299. Epub 2024 Dec 7 [PubMed PMID: 39644228]
Liu J, Jia J, Hao T, Ding J. Fahr's disease presenting with psychotic symptoms at onset in an adolescent: a case report. Neurocase. 2025 Aug:31(4):181-187. doi: 10.1080/13554794.2025.2522642. Epub 2025 Jun 23 [PubMed PMID: 40549752]
Level 3 (low-level) evidenceLi J, Wang X, Guo Y, Wang W, Wu S, Sun J. Fahr Syndrome, Hypoparathyroidism and Mitochondrial Encephalomyopathy With Lactic Acidosis and Stroke-Like Episodes (MELAS) Syndrome. AACE endocrinology and diabetes. 2026 Jan-Feb:13(1):102-106. doi: 10.1016/j.aed.2025.09.007. Epub 2025 Sep 22 [PubMed PMID: 41641297]
Wang X, Xu T, Zhu Y, Duan X. Fahr's Syndrome with Pseudohypoparathyroidism: Oral Features and Genetic Insights. International journal of molecular sciences. 2024 Oct 29:25(21):. doi: 10.3390/ijms252111611. Epub 2024 Oct 29 [PubMed PMID: 39519162]
Larsen TA, Dunn HG, Jan JE, Calne DB. Dystonia and calcification of the basal ganglia. Neurology. 1985 Apr:35(4):533-7 [PubMed PMID: 3982639]
Weisman DC, Yaari R, Hansen LA, Thal LJ. Density of the brain, decline of the mind: an atypical case of Fahr disease. Archives of neurology. 2007 May:64(5):756-7 [PubMed PMID: 17502478]
Level 3 (low-level) evidencePuvanendran K, Wong PK. Idiopathic familial basal ganglia calcification associated with juvenile hypertension. Journal of neurology, neurosurgery, and psychiatry. 1980 Mar:43(3):288 [PubMed PMID: 7373329]
Level 3 (low-level) evidenceBatla A, Tai XY, Schottlaender L, Erro R, Balint B, Bhatia KP. Deconstructing Fahr's disease/syndrome of brain calcification in the era of new genes. Parkinsonism & related disorders. 2017 Apr:37():1-10. doi: 10.1016/j.parkreldis.2016.12.024. Epub 2016 Dec 27 [PubMed PMID: 28162874]
Moskowitz MA, Winickoff RN, Heinz ER. Familial calcification of the basal ganglions: a metabolic and genetic study. The New England journal of medicine. 1971 Jul 8:285(2):72-7 [PubMed PMID: 4326703]
Ellie E, Julien J, Ferrer X. Familial idiopathic striopallidodentate calcifications. Neurology. 1989 Mar:39(3):381-5 [PubMed PMID: 2927646]
Level 3 (low-level) evidenceSnijders BMG, Koek HL, Peters MJL, Mali WPTM, van Beek MM, Betman MJC, Golüke NMS, Kruyswijk T, de Lange SV, Lith BDWT, Pekelharing RM, Roos MJ, Rutgers DR, Venema SMU, Verberne WR, Emmelot-Vonk MH, de Jong PA. Inter- and Intrarater Agreement of CT Brain Calcification Scoring in Primary Familial Brain Calcification. AJNR. American journal of neuroradiology. 2025 Jan 8:46(1):147-152. doi: 10.3174/ajnr.A8446. Epub 2025 Jan 8 [PubMed PMID: 39134371]
Alrashidi FS. Basal Ganglia Calcifications for Nephrologists: Fahr/Primary Familial Brain Calcification (PFBC) at the Interface of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) and Hypoparathyroidism. Cureus. 2025 Dec:17(12):e99031. doi: 10.7759/cureus.99031. Epub 2025 Dec 12 [PubMed PMID: 41531582]
Al Hariri B, Ali AA, Hassan AA, Abker SA, Satti AS, Gadkarem M, Mohammed OH, Nashwan AJ. Basal Ganglia Calcifications With Acute Behavioral Changes: A Case of Fahr's Syndrome. Cureus. 2025 Nov:17(11):e97802. doi: 10.7759/cureus.97802. Epub 2025 Nov 25 [PubMed PMID: 41458728]
Level 3 (low-level) evidenceSieffien W, Sorial S, Ure R, Golts M. Psychosis in Fahr Syndrome: A Case Report. The journal of ECT. 2025 Mar 1:41(1):e1-e3. doi: 10.1097/YCT.0000000000001048. Epub 2024 Jul 5 [PubMed PMID: 38968436]
Level 3 (low-level) evidenceZhao M, Cheng X, Chen L, Zeng YH, Lin KJ, Li YL, Zheng ZH, Huang XJ, Zuo DD, Guo XX, Guo J, He D, Liu Y, Lin Y, Wang C, Lv WQ, Su HZ, Yao XP, Ye ZL, Chen XH, Lu YQ, Huang CW, Yang G, Zhang YX, Lin MT, Wang N, Xiong ZQ, Chen WJ. Antisense oligonucleotides enhance SLC20A2 expression and suppress brain calcification in a humanized mouse model. Neuron. 2024 Oct 9:112(19):3278-3294.e7. doi: 10.1016/j.neuron.2024.07.013. Epub 2024 Aug 8 [PubMed PMID: 39121859]
Ebrahimi H, Holakoo A, Hejazian SE, Ahmadian H, Zohrevand AH, Sharifzadeh M. Incidental Diagnosis of Fahr's Disease Following Severe Traumatic Brain Injury: A Case Report. Clinical case reports. 2026 Jan:14(1):e71675. doi: 10.1002/ccr3.71675. Epub 2025 Dec 29 [PubMed PMID: 41473875]
Level 3 (low-level) evidencedi Biase L, Munhoz RP. Deep brain stimulation for the treatment of hyperkinetic movement disorders. Expert review of neurotherapeutics. 2016 Sep:16(9):1067-78. doi: 10.1080/14737175.2016.1196139. Epub 2016 Jun 10 [PubMed PMID: 27254274]
BRUYN GW, BOTS GT, STAAL A. FAMILIAL BILATERAL VASCULAR CALCIFICATION IN THE CENTRAL NERVOUS SYSTEM. Psychiatria, neurologia, neurochirurgia. 1964 Jul-Aug:67():342-76 [PubMed PMID: 14207403]
Zisimopoulou V, Siatouni A, Tsoukalos G, Tavernarakis A, Gatzonis S. Extensive bilateral intracranial calcifications: a case of iatrogenic hypoparathyroidism. Case reports in medicine. 2013:2013():932184. doi: 10.1155/2013/932184. Epub 2013 Feb 24 [PubMed PMID: 23509468]
Level 3 (low-level) evidenceEl Jabbour T, Aboursheid T, Keifo MB, Maksoud I, Alasmar D. Kenny-Caffey syndrome type 1. Avicenna journal of medicine. 2014 Jul:4(3):74-6. doi: 10.4103/2231-0770.133340. Epub [PubMed PMID: 24982829]
Level 3 (low-level) evidenceFinsterer J, Kopsa W. Basal Ganglia calcification in mitochondrial disorders. Metabolic brain disease. 2005 Sep:20(3):219-26 [PubMed PMID: 16167199]
Level 2 (mid-level) evidenceVerulashvili IV, Glonti LSh, Miminoshvili DK, Maniia MN, Mdivani KS. [Basal ganglia calcification: clinical manifestations and diagnostic evaluation]. Georgian medical news. 2006 Nov:(140):39-43 [PubMed PMID: 17179586]
Takashima S, Becker LE. Basal ganglia calcification in Down's syndrome. Journal of neurology, neurosurgery, and psychiatry. 1985 Jan:48(1):61-4 [PubMed PMID: 3156213]
Ukai K, Kosaka K. Diffuse neurofibrillary tangles with calcification (Kosaka-Shibayama disease) in Japan. Psychiatry and clinical neurosciences. 2016 Mar:70(3):131-40. doi: 10.1111/pcn.12334. Epub 2015 Aug 19 [PubMed PMID: 26176797]
Chandrasekaran S, Nanjundan M, Natarajan S, Ramadhas K. Radiologic presentation of lipoid proteinosis with symmetrical medial temporal lobe calcifications. Radiology case reports. 2015:10(2):1121. doi: 10.2484/rcr.v10i2.1121. Epub 2016 Feb 17 [PubMed PMID: 27398129]
Level 3 (low-level) evidenceRevesz T, Fletcher S, al-Gazali LI, DeBuse P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? Journal of medical genetics. 1992 Sep:29(9):673-5 [PubMed PMID: 1404302]
Level 3 (low-level) evidenceFörstl H, Krumm B, Eden S, Kohlmeyer K. Neurological disorders in 166 patients with basal ganglia calcification: a statistical evaluation. Journal of neurology. 1992 Jan:239(1):36-8 [PubMed PMID: 1541967]
Ostling S, Andreasson LA, Skoog I. Basal ganglia calcification and psychotic symptoms in the very old. International journal of geriatric psychiatry. 2003 Nov:18(11):983-7 [PubMed PMID: 14618548]
Level 2 (mid-level) evidenceRaymond AA, Zariah AA, Samad SA, Chin CN, Kong NC. Brain calcification in patients with cerebral lupus. Lupus. 1996 Apr:5(2):123-8 [PubMed PMID: 8743125]
Level 2 (mid-level) evidenceSanaei E, Bidaki R, Pashmchi M, Jalalifard H. Challenging Diagnosis of Fahr's Disease Mimicking Parkinson's Disease: A Case Report. Clinical case reports. 2025 Feb:13(2):e70250. doi: 10.1002/ccr3.70250. Epub 2025 Feb 18 [PubMed PMID: 39973894]
Level 3 (low-level) evidenceBhinder KK, Khalid N, Akhtar S. Multi territory ischemic stroke in a patient with Fahr's disease: Report of a rare case. Radiology case reports. 2025 Jan:20(1):661-666. doi: 10.1016/j.radcr.2024.10.100. Epub 2024 Nov 9 [PubMed PMID: 39583223]
Level 3 (low-level) evidence