Introduction
Ehlers-Danlos syndromes (EDS) are a heterogeneous group of heritable connective tissue disorders marked by joint hypermobility, skin hyperextensibility, and tissue fragility.[1][2] Clinical presentation varies and may involve multiple organ systems, contributing to frequent underrecognition and delayed diagnosis.
The 2017 International Classification recognized 13 types of EDS with distinct clinical features, 12 of which are caused by variants in 20 genes.[3][4][3]The molecular basis of hypermobile EDS (hEDS), the most common type, remains unknown. Since 2017, a 14th type, the classical-like EDS type 2 (clEDS2), has been described, bringing the current count to 14, listed below:[2][5][6]
- Classical (cEDS): skin hyperextensibility, atrophic scarring, joint hypermobility
- Classical-like (clEDS): skin hyperextensibility, joint hypermobility, easy bruising
- Classical-like type 2 (clEDS2): skin fragility, joint hypermobility
- Cardiac-valvular (cvEDS): progressive valvular disease, connective tissue features
- Vascular (vEDS): arterial and organ fragility, the most severe subtype
- Hypermobile (hEDS): generalized joint hypermobility, chronic pain; most common type
- Arthrochalasia (aEDS): congenital hip dislocation, severe joint hypermobility
- Dermatosparaxis (dEDS): extreme skin fragility and redundancy
- Kyphoscoliotic (kEDS): hypotonia, progressive kyphoscoliosis
- Brittle cornea syndrome (BCS): corneal thinning and fragility
- Spondylodysplastic (spEDS): short stature, skeletal abnormalities
- Musculocontractural (mcEDS): congenital contractures, craniofacial features
- Myopathic (mEDS): muscle hypotonia with joint involvement
- Periodontal (pEDS): early-onset periodontitis, gingival abnormalities [2][5][6]
The underlying defect involves disrupted collagen biosynthesis, structure, or processing within the ECM. EDS affect nearly every organ system and often lead to significant health complications and reduced quality of life.[3][4] Diagnosis is primarily clinical, with genetic testing used when available. Accurate type identification is essential for risk stratification, management, and counseling because disease severity and complications vary widely.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
EDS results from pathogenic variants in at least 20 genes that disrupt the biosynthesis, processing, organization, or supramolecular assembly of collagen fibrils and other ECM components.[2] These variants disrupt collagen fibrillogenesis, cross-linking, or interactions with other matrix components, resulting in tissue fragility and multisystem manifestations. Causative genes include those encoding fibrillar collagens (eg, COL5A1, COL5A2, COL3A1, COL1A1, COL1A2), collagen-processing enzymes (eg, ADAMTS2, PLOD1, FKBP14), and other ECM components (eg, TNXB), as well as pathways involved in glycosaminoglycan synthesis and intracellular regulation.[7][5] The following specific gene mutations and inheritance patterns define EDS types:
- cEDS: COL5A1, COL5A2 (type V collagen regulating fibrillogenesis); rarely COL1A1 — autosomal dominant[8]
- clEDS
- Type 1: TNXB (tenascin-X, ECM glycoprotein); autosomal recessive
- Type 2: AEBP1 (facilitates collagen polymerization); autosomal recessive
- cvEDS: COL1A2 (absence of pro-α2 chains of type I collagen); autosomal recessive
- vEDS: COL3A1 (type III collagen in vessel walls); rarely COL1A1; autosomal dominant
- hEDS: Genetic basis unknown; presumed autosomal dominant
- aEDS: COL1A1, COL1A2 (defective N-propeptide cleavage of type I collagen); autosomal dominant
- dEDS: ADAMTS2 (procollagen I N-proteinase deficiency); autosomal recessive
- kEDS: PLOD1 (lysyl hydroxylase 1, collagen cross-linking) or FKBP14 (collagen chaperone); autosomal recessive
- BCS: ZNF469, PRDM5 (transcriptional regulation of ECM); autosomal recessive
- spEDS: B4GALT7, B3GALT6 (GAG linker synthesis), SLC39A13 (zinc transport); autosomal recessive
- mcEDS: CHST14, DSE (dermatan sulfate biosynthesis); autosomal recessive
- mEDS: COL12A1 (type XII collagen, fibril organization); autosomal dominant or recessive
- pEDS: C1R, C1S (complement pathway proteases); autosomal dominant [5]
Most EDS types, including hypermobile, classical, and vascular, follow an autosomal-dominant inheritance pattern and account for approximately 80% to 90% of cases in clinical practice.[2] Individuals with hypermobile EDS account for the vast majority of cases, contributing to the predominance of autosomal dominant inheritance. Autosomal recessive forms are rare.[2][3] De novo pathogenic variants occur in a substantial minority of cases, particularly in vascular EDS, where approximately 50% have no family history.[9][10][11]
Epidemiology
The overall prevalence of EDS is estimated at approximately 1 in 2,500 to 1 in 5,000 individuals, though this likely underestimates the true prevalence due to underrecognition and diagnostic challenges.[12] Prevalence varies by type, with hEDS accounting for approximately 80% to 90% of diagnoses. Estimated prevalence ranges from 1 in 3,100 to 1 in 20,000, reflecting variability in diagnostic criteria and the absence of a confirmed genetic marker.[13][14] Classical EDS is less common, affecting approximately 1 in 10,000 to 1 in 20,000 individuals, while vascular EDS is rare, with estimates ranging from 1 in 50,000 to 1 in 250,000.[15][16] All other subtypes are exceedingly rare, often described in only a small number of families worldwide.[10]
EDS occurs globally and affects individuals of all racial and ethnic groups, with no clear geographic or ethnic predilection. However, most epidemiologic data are derived from European and North American populations.[17] Diagnosed cohorts show a female predominance, particularly in hEDS, with reported female-to-male ratios ranging from approximately 1.4:1 overall to 70% to 90% female in hEDS.[15][14] Whether this reflects true sex-related differences in disease expression, eg, hormonal influences on connective tissue laxity, or ascertainment bias related to differences in healthcare-seeking behavior, symptom reporting (eg, pain, fatigue), and referral patterns, remains unclear.[18]
The mean age at diagnosis is approximately 35 years, although symptoms often begin earlier, and delays in diagnosis are common.[15] Despite growing recognition, the true prevalence of individual EDS types remains uncertain because many cases, particularly milder forms, may go undiagnosed.[19][20]
Pathophysiology
The pathophysiology of EDS is primarily due to pathogenic variants in genes involved in the biosynthesis, processing, organization, or extracellular assembly of collagen and other ECM components.[21] Because collagen is the most abundant protein in the body and is widely distributed throughout the skin, blood vessels, joints, viscera, and musculoskeletal system, defects in collagen structure or processing produce multisystem manifestations by disrupting the structural integrity of connective tissue throughout the body. The specific clinical phenotype depends on which collagen type is affected and on its tissue distribution.[5]
Collagen fibrils normally form through tightly regulated intracellular synthesis of procollagen, followed by extracellular processing and assembly into highly ordered fibrils. Pathogenic variants, particularly those affecting conserved glycine residues within the Gly-X-Y repeat, disrupt triple-helical formation and fibrillogenesis, resulting in structurally abnormal or deficient collagen.
Disease mechanisms are broadly categorized as defects in fibrillar collagen genes (eg, COL3A1, COL5A1, COL5A2, COL1A1, COL1A2), defects in collagen-processing enzymes (eg, lysyl hydroxylase and related modifying enzymes), and defects in other extracellular matrix components or interacting proteins (eg, tenascin-X).[5][21] These abnormalities impair collagen cross-linking, fibril formation, and matrix stability. Downstream effects include heightened cellular stress responses, such as endoplasmic reticulum stress, impaired autophagy, and abnormal cell–matrix signaling, which contribute to tissue fragility and inflammation.[22]
Because collagen is widely distributed in skin, blood vessels, ligaments, and hollow organs, clinical manifestations reflect multisystem connective tissue weakness. Tissue-specific expression of different collagen types contributes to phenotypic variability. For example, type III collagen predominates in vascular structures, which may explain the risk of arterial and organ rupture in vascular EDS. Overall, pathogenic variants impair collagen fibrillogenesis and weaken the extracellular matrix architecture, resulting in shared features of skin hyperextensibility, poor wound healing, joint hypermobility, and tissue fragility, with severity dependent on the EDS type and specific mutation.[2]
History and Physical
Clinical presentation in EDS varies by type and severity, but several characteristic features commonly prompt clinical evaluation. Because EDS results from inherited defects in collagen structure, processing, or extracellular matrix integrity, the underlying abnormality is present at birth. However, manifestations may not become clinically apparent until childhood or later. Common presenting features include skin hyperextensibility, joint hypermobility, easy bruising, chronic musculoskeletal pain, recurrent sprains, subluxations, and dislocations.[2]
Clinical History
A detailed patient history includes assessment of the following symptoms involving the skin, musculoskeletal system, vasculature, gastrointestinal tract, and pelvic floor, as well as a family history:
- Skin: Symptoms may include hyperextensible or unusually soft skin, delayed wound healing, widened or atrophic scars, easy bruising, recurrent lacerations, and poor tissue strength.
- Musculoskeletal: Clinical symptoms include generalized or localized joint hypermobility, recurrent sprains, atraumatic dislocations (commonly shoulder or patella), chronic pain, early fatigue, muscle weakness, frequent falls, impaired coordination, and early osteoarthritis. Congenital hip dislocation may occur in some types.[23] Patients may report being "double-jointed" or having extreme flexibility since childhood.[24]
- Autonomic and functional complaints: Patients may exhibit orthostatic intolerance, fatigue, exercise intolerance, and reduced postural control, particularly in children with hypermobile forms of EDS.[25]
- Gastrointestinal and pelvic: Symptoms include hernias, rectal prolapse, constipation, diarrhea, urinary incontinence, and pelvic organ prolapse.[24]
- Dental: Patients may exhibit delayed tooth eruption, hypodontia, dentin abnormalities, gingival fragility, or periodontal disease.[26][27]
- Vascular or visceral events: Patients may have an arterial aneurysm, dissection, rupture, spontaneous pneumothorax, bowel perforation, uterine rupture, or unexplained severe bruising, which raises concern for vascular EDS.
- Pregnancy: Patients may report prior uterine rupture, severe peripartum hemorrhage, tissue tearing, or vascular complications.
- Developmental history (pediatric patients): These types of symptoms include motor delay, clumsiness, or speech delay, particularly in hypermobile EDS.[24]
- Family history: Patients may report relatives with hypermobility, abnormal scarring, arterial rupture, sudden unexplained death, recurrent dislocations, or a known EDS diagnosis. In hypermobile EDS, manifestations in relatives may be subtle and require directed questioning.[24]
Physical Examination Findings
Skin findings
The skin may be soft, hyperextensible, and recoil promptly after release, which helps distinguish EDS from other causes of skin laxity. Skin hyperextensibility can be assessed by gentle stretching; abnormal values include more than 1.5 cm on the forearm or dorsum of the hand, and more than 3 cm at the neck, elbows, or knees. Atrophic or widened ("cigarette paper") scars from delayed wound healing are characteristic of some EDS types. Visible subcutaneous veins and translucent skin suggest vascular EDS. Molluscoid pseudotumors (fleshy lesions over scars or pressure points) and subcutaneous spheroids (small, mobile, calcified nodules, often over the forearms or shins) may be seen in classical EDS.[18][28]
Joint and musculoskeletal findings
The Beighton score is a 9-point physical examination tool used to assess generalized joint hypermobility (see Image. Beighton Criteria). This tool evaluates passive dorsiflexion of the fifth fingers, thumb apposition to the forearm, elbow, and knee hyperextension greater than 10°, and forward trunk flexion with palms flat on the floor. A score of 5/9 or higher in adults and 6/9 or more in children is considered positive. Because laxity typically decreases with age, the 5-point questionnaire (5PQ) supplements the Beighton score in individuals with acquired joint limitation.[29] Affected individuals may exhibit pes planus, scoliosis, muscle weakness, and gait abnormalities. Small joints of the hands are frequently involved, although any joint may be affected.
Cardiovascular findings
Auscultation for valvular disease is particularly important in cardiac-valvular EDS. Blood pressure, pulse asymmetry, bruits, or signs of arterial disease should prompt urgent vascular assessment.
Developmental (pediatric) considerations
In infants and children, motor development should be carefully assessed, as clumsiness or motor delay may support the diagnosis, particularly in hEDS.[24]
Physical Examination Type-Specific Clues
Type-specific physical exam findings include:
- Classical EDS: marked skin hyperextensibility, atrophic scarring, generalized joint hypermobility, easy bruising, molluscoid pseudotumors, subcutaneous spheroids, and hernias
- Classical-like EDS: skin hyperextensibility without atrophic scarring, joint hypermobility, easy bruising, possible mild muscle weakness, neuropathy, or foot deformities
- Hypermobile EDS: generalized joint hypermobility with chronic pain, instability, fatigue, and associated functional symptoms [24]
- Vascular EDS: thin translucent skin, characteristic bruising, arterial or organ rupture, spontaneous pneumothorax, carotid-cavernous fistula, or pregnancy-related uterine rupture
- Cardiac-valvular EDS: skin and joint findings with progressive valvular heart disease
Because no single laboratory test confirms most EDS types, diagnosis depends on recognizing characteristic history and examination findings, followed by targeted genetic evaluation when appropriate.
Evaluation
General Diagnostic Approach
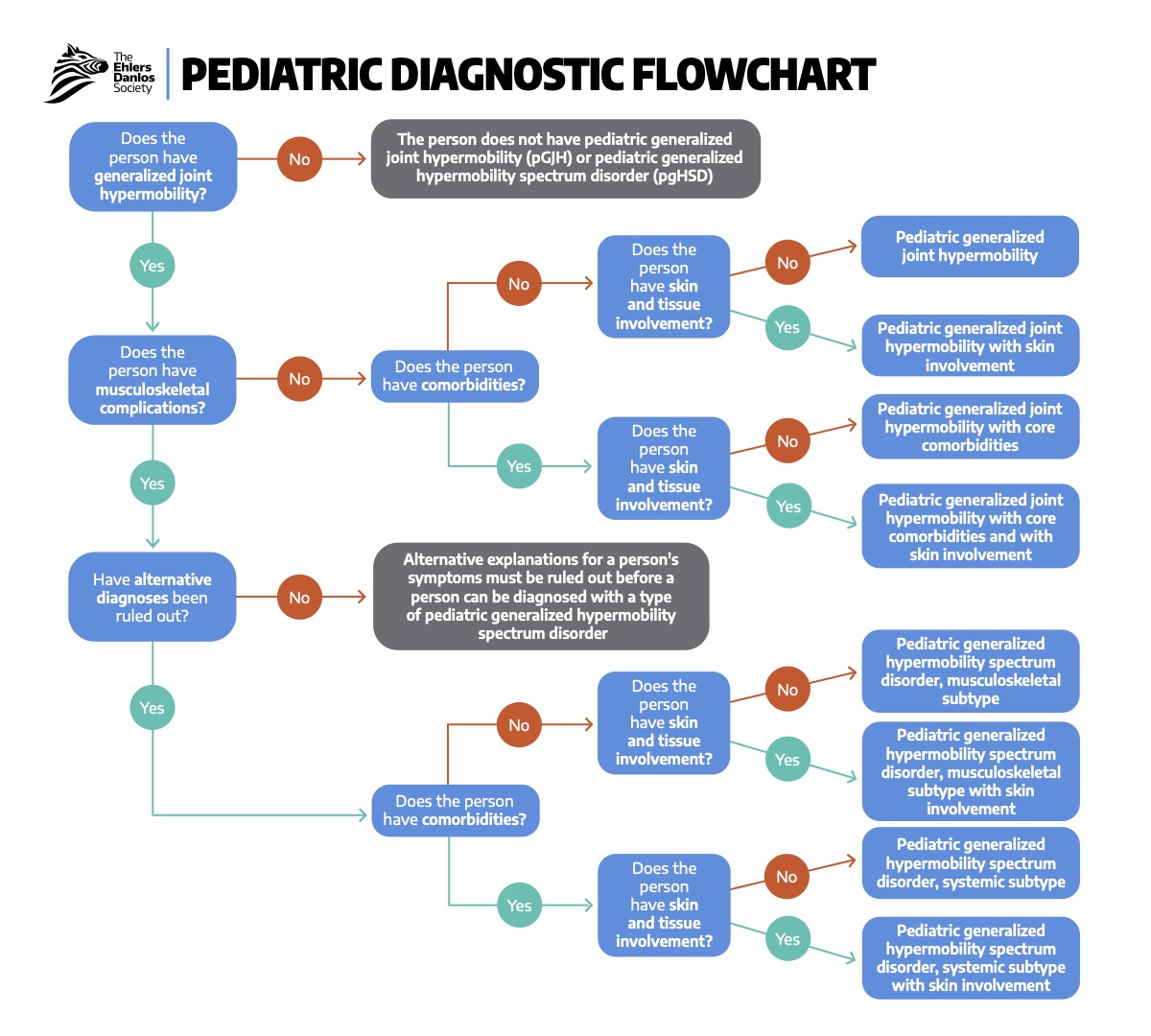
The evaluation of EDS begins with clinical suspicion based on characteristic features, followed by targeted evaluation and molecular genetic testing to confirm the diagnosis and determine the type (see Image. Pediatric Diagnostic Flowchart).[30] In contrast, hEDS (the most common type) remains a clinical diagnosis because no causative gene has been identified.[31] The workup should also assess the extent of organ-system involvement and any associated complications.
Type-Specific Diagnostic Approach
The following criteria are utilized to diagnose specific types of EDS:
- hEDS: All 3 of the following criteria must be present for diagnosis.[31][24]
- Generalized joint hypermobility. The hEDS type is the only type that uses the Beighton score as a formal component of its diagnostic criteria (see Image. Beighton Criteria)
- Systemic manifestations of a connective tissue disorder or musculoskeletal complications
- Exclusion of alternative diagnoses; genetic testing may reveal an alternative etiology in a substantial proportion of patients who initially meet hEDS criteria.[31][24]
- cEDS: Diagnosis requires skin hyperextensibility with atrophic scarring (major criterion), plus generalized joint hypermobility or at least 3 minor criteria. Molecular confirmation involves variants in COL5A1, COL5A2, or COL1A1.[8]
- vEDS: Definitive diagnosis requires identification of a pathogenic variant in COL3A1. Clinical features alone have limited sensitivity, particularly in younger patients without a family history.[7][32]
Genetic Testing
Molecular confirmation is the diagnostic standard for all EDS types except hEDS.[5][2] Clinical features associated with a higher likelihood of a positive molecular diagnosis include joint hypermobility, skin hyperextensibility, poor wound healing, easy bruising, atrophic scarring, and developmental dysplasia of the hip.[30]
Multigene panels are the preferred first-line test and include genes, eg, COL5A1, COL5A2, COL3A1, COL1A1, TNXB, PLOD1, and AEBP1. Diagnostic yield is high in patients meeting clinical criteria.[30] Single-gene testing may be appropriate when a specific subtype is strongly suspected (eg, COL3A1 testing in suspected vEDS). Whole exome sequencing or chromosomal microarray may be considered when panel testing is negative, but suspicion remains high.[30]
Adjunct Investigations
The following diagnostic studies may be used to evaluate patients for conditions associated with EDS:
- Cardiovascular evaluation
- A baseline transthoracic echocardiogram is particularly useful in classical EDS and in children younger than 10.[9]
- If initial imaging is normal, routine serial imaging may not be necessary in classical EDS.
- In vascular EDS, periodic vascular surveillance with Doppler ultrasound, computed tomography angiography (CTA), or MR angiography (MRA) is recommended.[7]
- Imaging
- Laboratory studies
- Routine bloodwork is generally not diagnostic.
- Coagulation studies are typically normal, even in patients with easy bruising, unless another bleeding disorder is present.[9]
- Evaluation of clotting factors may be considered when bleeding symptoms are prominent to exclude comorbid conditions.
- Rare associations, eg, hemophilia A in vascular EDS, have been reported.[35]
- Histologic and ultrastructural analysis: A skin biopsy examined by transmission electron microscopy may demonstrate characteristic abnormalities in collagen structure (eg, "collagen flowers" in classical EDS). This modality is used infrequently due to the availability of genetic testing.[9][2][23]
- Biochemical testing: Analysis of type III procollagen in cultured fibroblasts supports a diagnosis of vascular EDS, but has largely been replaced by molecular testing.[36]
Symptom-Directed Testing
Further evaluation should be guided by presenting symptoms. For example, if the cardiovascular workup is negative in a patient with suspected EDS and pre-syncope, a tilt table test may be ordered to evaluate for an associated syndrome, eg, postural orthostatic tachycardia syndrome (POTS) or another form of dysautonomia.[37][38]
Treatment / Management
Management of EDS is interprofessional and primarily supportive, as no cure exists. Care focuses on symptom control, preventing complications, and surveillance for organ involvement, with strategies tailored to the specific EDS type.[9][5]
General Principles
Care is best coordinated by a primary clinician, with referrals to specialists, eg, genetics, cardiology, orthopedics, pain management, and high-risk obstetrics, as needed.[5] Preventing injuries and complications is central to management, including the following areas:
- Physical and occupational therapy: These therapies are the cornerstones of treatment and may improve pain, motor function, proprioception, and quality of life, although evidence from randomized clinical trials is limited.[39]
- Targeted exercise programs to increase core and extremity strength, joint stability, and proprioception
- Motor function training and low-impact exercise programs
- Bracing, splints, and orthoses for joint support
- Assistive devices, eg, wheelchairs and wide-grip writing utensils, and ergonomic adaptations [40][41]
(B2)
- Pain management: Chronic pain is common and often requires a multimodal approach.[42][41]
- First-line: physical therapy, behavioral therapy (including cognitive behavioral therapy), and activity modification
- Pharmacologic options: acetaminophen and NSAIDs; opioids (effective but should be used with extreme caution)
- Neuropathic agents (eg, gabapentin, duloxetine, tricyclic antidepressants) have variable efficacy and tolerability.
- Skin and wound care: Due to impaired collagen integrity, patients are at increased risk of poor wound healing and dehiscence. Management strategies aim to prevent dehiscence.[23][43]
- Cardiovascular management and surveillance: Regular cardiovascular monitoring is recommended to identify and mitigate complications.
- Screening echocardiography for aortic root dilation or valvular disease [34]
- Aggressive management of hypertension to reduce vascular stress
- Celiprolol, a third-generation beta-blocker with vasodilatory properties, has demonstrated benefit in reducing arterial dissections, aneurysms, and rupture in patients with vascular EDS. However, because this agent is not approved by the United States Food and Drug Administration, alternative beta-blockers are often prescribed.[45]
(A1)
Type-Specific Considerations
Some EDS types have the following targeted management recommendations:
- hEDS: Management of hEDS is largely supportive, addressing associated comorbidities.[41]
- Autonomic dysfunction (eg., POTS): increased fluid and salt intake, compression garments, graded exercise; medications such as fludrocortisone or midodrine for refractory symptoms [46]
- Gastrointestinal symptoms: standard therapies for reflux, dysmotility, constipation, or diarrhea
- Mast cell activation symptoms: antihistamines and adjunctive therapies
- Psychological support: management of anxiety, depression, and functional symptoms
- cEDS
- Emphasis on skin protection and wound care
- Baseline echocardiogram, with follow-up guided by findings
- Avoidance of high-impact or high-strain activities
- vEDS: This type of EDS requires intensive surveillance and management.
- Baseline head-to-pelvis vascular imaging (CTA or MRA), with annual or biannual follow-up depending on findings [47]
- Recognition that arterial events may occur without significant dilation
- Beta-blocker therapy
- Surgical intervention carries an increased risk due to vascular fragility
Surgical Considerations
Surgical intervention should remain limited to cases in which conservative management proves unsuccessful. Tissue fragility, impaired wound healing, and increased susceptibility to infection frequently compromise surgical outcomes. Procedures require experienced interprofessional teams familiar with EDS-specific perioperative risks. Careful planning and monitoring remain essential, particularly regarding anesthesia management, patient positioning, and bleeding risk.[48]
Pregnancy Considerations
Pregnancy in patients with EDS requires management by obstetricians experienced in high-risk maternal care.[43] In vascular EDS, major maternal complications include uterine rupture and serious vascular events. Planned cesarean delivery at 37 weeks of gestation often forms part of the management strategy. Patients with hEDS and cEDS frequently experience pregnancy-related complications; however, outcomes generally remain favorable with appropriate monitoring and interprofessional care.[49]
Long-term Surveillance for All Types
Long-term surveillance across all EDS types should include regular assessment of joint stability, chronic pain, and functional status. Ongoing monitoring of cardiovascular health remains particularly important in patients with vEDS. Additional follow-up should address bleeding tendencies, wound healing complications, and gastrointestinal, autonomic, and psychological symptoms.
Differential Diagnosis
Connective Tissue Disorders
The differential diagnosis requires distinguishing among the 13 recognized types, distinguishing EDS from hypermobility spectrum disorders, and excluding other heritable connective tissue disorders with overlapping phenotypes. Hypermobility spectrum disorders are diagnosed in individuals with symptomatic joint hypermobility who do not meet all 3 hEDS criteria and who have no alternative diagnosis.[3] Growing evidence suggests hEDS and hypermobility spectrum disorders may represent a phenotypic continuum rather than distinct entities.[50]
Significant clinical overlap exists among EDS types and other connective tissue disorders, including Marfan syndrome, Loeys-Dietz syndrome, and osteogenesis imperfecta, making molecular confirmation essential. Notably, 13.9% of patients clinically diagnosed with classical EDS have been found to harbor pathogenic variants in genes other than COL5A1 or COL5A2.[8]
Osteogenesis imperfecta
Osteogenesis imperfecta type I may be confused with EDS because of shared features, including joint hypermobility and significant morbidity after minor trauma. The distinction relies on clinical findings in conjunction with genetic testing. Patients with non-deforming osteogenesis imperfecta (type I) commonly exhibit blue sclerae, sensorineural hearing loss, wormian bones, and dentinogenesis imperfecta. Blue sclerae may also be observed in EDS, and both conditions can involve mutations in COL1A1 and COL1A2. Other OI types are generally not included in the differential diagnosis.[51]
Marfan syndrome
Marfan syndrome shares several features with EDS, particularly the hypermobile type; however, Marfan syndrome can typically be distinguished clinically and genetically.[9][2] Marfan syndrome is most commonly associated with mutations in the FBN1 gene, which encodes fibrillin-1. Patients may present with joint hypermobility, marfanoid habitus, ectopia lentis, and aortic root pathology. Although both conditions may involve aortic root dilation, progression over time is more typical of Marfan syndrome than of EDS. Additionally, elbow hypermobility is less common, and individuals with Marfan syndrome often have an arm span-to-height ratio greater than 1.05. Genetic testing that identifies FBN1 mutations supports the diagnosis of Marfan syndrome and distinguishes it from EDS.[9][51]
Loeys-Dietz syndrome
Loeys-Dietz syndrome can resemble EDS due to its autosomal dominant inheritance and early-onset vascular complications, particularly aneurysms. However, Loeys-Dietz syndrome is characterized by a clinical triad of bifid uvula or cleft palate, hypertelorism, and widespread arterial aneurysms.[51][52] Unlike EDS, in which vascular involvement may be more localized (eg, the aortic root), aneurysms in Loeys-Dietz syndrome tend to involve multiple arterial beds.
Cutis laxa
Cutis laxa may also be mistaken for EDS on initial examination. Differentiation is primarily based on skin characteristics: although EDS types feature hyperextensible skin, it typically recoils immediately after release, whereas skin in cutis laxa returns slowly after distention. Both conditions may involve the cardiovascular system and lead to hernia formation. Genetic testing in cutis laxa often reveals mutations in genes, eg, FBLN5 (fibulin-5).[9][51][53]
Additional Differential Diagnoses
Patients with EDS, particularly the hypermobile type, are frequently misdiagnosed with fibromyalgia, chronic fatigue syndrome, or depression due to overlapping symptoms and associated psychosocial burden.[9][20][54] Although these conditions may coexist, clinicians should maintain a high index of suspicion to avoid missed or delayed diagnosis of EDS and its associated complications.
Special consideration is warranted for patients with suspected EDS presenting after trauma, as they are at increased risk of joint dislocation and vascular and visceral injury. Connective tissue disorders should be considered in younger patients with examination findings disproportionate to the reported mechanism of injury. In children, injuries that are out of proportion to the stated history should also raise concern for non-accidental trauma and prompt appropriate evaluation.[55]
Prognosis
Prognosis varies by type and is influenced by clinical severity and environmental factors. Overall outcomes range from near-normal life expectancy with chronic morbidity to substantially reduced survival in more severe forms. Early diagnosis, patient education, and avoidance of high-risk activities can mitigate morbidity, particularly in hEDS.[20]
Mortality varies markedly among EDS types. Hypermobile and classical EDS are generally not associated with reduced life expectancy, though both can lead to significant long-term morbidity. Cutaneous and joint manifestations are common and may progress with age, but life-threatening complications are less frequent, and overall survival is typically preserved.[28] Hypermobile EDS is not associated with increased mortality but is marked by substantial chronic morbidity. Patients frequently experience persistent pain, fatigue, and functional impairment, which significantly reduce quality of life. Reported outcomes indicate disability and symptom burden comparable to or exceeding those seen in other chronic conditions.[56][57][6]
Vascular EDS carries the most serious prognosis due to the risk of early, life-threatening vascular events. Historically, median life expectancy in vEDS has been estimated at approximately 48 years; however, more recent observational data suggest improved outcomes, with survival rates of 99.3% at 1 year, 89.9% at 5 years, and 83.4% at 10 years following molecular diagnosis.[58][59][60] Kyphoscoliotic EDS is also associated with reduced life expectancy, largely due to cardiopulmonary involvement.[61]
Across all EDS types, population-level data suggest a reduced average lifespan relative to the general population, with a reported mean age at death of 53.6 years. Socioeconomic impact is also notable, including lower educational attainment and increased rates of early retirement and disability support.[60][58]
Beyond physical health, EDS are associated with a significant psychosocial burden. The chronic nature of the symptoms, absence of curative therapy, and variable response to treatment may lead to frustration, reduced trust in healthcare systems, and decreased care engagement over time. Patients also report diminished quality of life related to chronic pain, functional limitations, and a perceived lack of validation when objective findings do not accompany symptoms.[9][54]
Complications
Organ System Complications
Complications affect multiple organ systems and vary by type, with vascular complications being the most life-threatening. A recent meta-analysis reported an overall pooled prevalence of vascular complications of 30% across EDS types, with the highest rates observed in vEDS (42.4%), followed by hEDS (19.8%) and unspecified EDS (18.7%).[62] Arterial dissection and rupture are the most serious complications and the leading cause of death, occurring in up to 35.5% of patients with vEDS and accounting for approximately 70.6% of deaths in one cohort.[60][63][6] Aneurysm formation (31.4%) and cerebrovascular events (16.3%) are also reported.[6] Vascular rupture can occur throughout the arterial system, most commonly in the thorax and abdomen.[9][2] Approximately 25% of patients with vEDS experience a first major complication by age 20, and more than 80% by age 40.[58]
Visceral complications arise from underlying tissue fragility and include spontaneous or trauma-related rupture of both hollow and solid organs. The most commonly affected sites include the sigmoid colon, gravid uterus, spleen, and liver.[59][63] Gastrointestinal involvement includes bowel perforation, which accounts for approximately 25% of major complications in vEDS, as well as functional gastrointestinal disorders that are highly prevalent in hEDS (up to 90.5%).[58][6] Hernias and rectal prolapse are also common.[9][23] Pulmonary complications include pneumothorax and hemothorax, particularly in vEDS and kyphoscoliotic EDS.[63][61] Sudden onset of dyspnea in a patient with known or suspected EDS should prompt emergent evaluation.[2][64]
Musculoskeletal complications are the most prevalent across EDS types and contribute significantly to morbidity. These include chronic joint pain (80.4%), decreased bone density (90.7%), hypotonia or muscle weakness (56.4%), recurrent joint dislocations and subluxations, and early-onset progressive osteoarthritis.[6] Recurrent joint instability may lead to repeated injuries and the need for orthopedic interventions, although outcomes may be limited by underlying connective tissue laxity. Kyphoscoliosis, characteristic of the kyphoscoliotic type, may impair pulmonary function.[65]
Cardiac involvement most commonly includes mitral valve prolapse, which may occur with or without regurgitation.[9][2][34] Although often mild, regurgitation can become clinically significant under physiologic stress, eg, pregnancy, and may present with heart failure or pulmonary edema.[34][43]
Obstetric Complications
Obstetric complications are increased across EDS types due to tissue and vascular fragility. Patients are at higher risk for preterm delivery (OR 1.35), cervical insufficiency (OR 2.14), postpartum hemorrhage (OR 1.41), cesarean delivery (OR 1.26), and severe maternal morbidity (OR 1.84).[66] Additional complications include premature rupture of membranes, precipitous labor, breech presentation, uterine and bladder prolapse, and increased risk of vaginal and perineal lacerations.[9][23][43] In vEDS, pregnancy is associated with significant risk, including uterine rupture, arterial rupture, and maternal mortality of approximately 5.7% per pregnancy.[67]
Across EDS types, preterm birth occurs in approximately 25%, spontaneous abortion in 57%, and infertility is reported in 44%.[68] Due to vascular fragility, patients have an increased risk of bleeding compared to the general population. Preexisting cardiovascular conditions, such as mitral valve prolapse or aortic dilation, should be monitored closely during pregnancy. Birth injuries to both the fetus and mother are more common, including dislocations for the child and higher-grade injuries to the laboring mother. Despite these increased risks, it is unclear whether delivery via planned cesarean section over vaginal delivery to the patient or newborn is advantageous.[43]
Additional Complications
Autonomic dysfunction is frequently observed, particularly in hEDS. POTS is reported in up to 58.3% of patients and may present with palpitations, dizziness, or syncope.[56][37][38] Mast cell activation syndrome (MCAS) has also been described (32.1%), and the combination of hEDS, POTS, and MCAS occurs in approximately 25% of patients.[56] Orthostatic intolerance is common and may reflect benign autonomic dysfunction; however, in the acute setting, it should prompt consideration of hypovolemia or vascular injury.
Sleep-related disorders, including obstructive sleep apnea, are associated with EDS and may contribute to chronic fatigue. Craniofacial and soft tissue abnormalities are thought to underlie this relationship.[2][69] Additionally, surgical and procedural interventions carry increased risk due to vascular fragility, increased bleeding tendency, and delayed wound healing. These factors should be carefully considered when planning both elective and emergent procedures.[23][20] Given the risk of life-threatening complications, the sudden onset of severe pain in a patient with known or suspected EDS should prompt emergent medical evaluation, even in the absence of trauma.
Deterrence and Patient Education
The cornerstone of management is patient education focused on injury prevention, risk reduction, and setting realistic expectations for disease progression. Patients should be counseled about the chronic nature of the condition and the importance of long-term adherence to recommended therapies, including physical therapy and activity modification.[9][20][65]
Avoiding high-risk activities is essential to reducing the risk of joint injury and long-term musculoskeletal complications. Patients should be advised to limit or avoid contact sports, heavy weightlifting, and other high-impact or high-resistance activities. Instead, low-impact and low-resistance exercises are encouraged, as they may help maintain joint stability and reduce morbidity.[65] Therapeutic exercise and motor function training have shown benefit, particularly for patients with hypermobile EDS and hypermobility spectrum disorders.[39] Because joint instability predisposes to recurrent subluxations and dislocations, preventing initial and repeat injuries is critical, as each event increases the risk of chronic pain and early-onset osteoarthritis.[9][2][20]
Patients with vascular involvement or at risk for vascular complications should be counseled to adhere strictly to antihypertensive therapy, including beta-blockers when indicated, as this may reduce the risk of future vascular events.[59]
Genetic counseling is an essential part of patient education. In vEDS, the relatively high rate of de novo mutations has significant implications: a negative family history does not rule out the diagnosis. Patients and families should be counseled about inheritance patterns, variability in disease expression, and the possibility of new mutations even when parents are unaffected.[11]
Enhancing Healthcare Team Outcomes
Patients with EDS benefit from a coordinated interprofessional approach due to the disorder's multisystem nature. Management usually begins with frontline clinicians, including physicians, nurse practitioners, and physician assistants, who must maintain a high index of suspicion to establish an accurate diagnosis. Evaluation is based on a detailed personal and family history, a comprehensive physical examination, and, when appropriate, genetic testing.[23] Because no single standardized management algorithm exists, care is typically individualized, with referral to appropriate specialists guided by the patient's specific manifestations and risk of complications.
A knowledgeable primary care clinician plays a central role in coordinating care, monitoring for complications, and ensuring continuity of care across specialties. Depending on the extent of disease involvement, the care team may include cardiology, vascular surgery, orthopedics, physical and occupational therapy, pain management, gastroenterology, obstetrics/gynecology, and mental health professionals.[9][20][41] Patient education is a critical component of effective management. Individuals should be counseled to recognize warning signs of serious complications and to seek prompt medical attention when indicated. Patients with vascular involvement are often advised to carry medical-alert identification to facilitate rapid recognition and appropriate emergency care.[70]
Genetic counseling is essential for patients and families. Genetic testing can help determine the specific EDS type and inheritance pattern, informing prognosis and reproductive decision-making.[9] The relatively high rate of de novo mutations, particularly in vascular EDS, should be discussed, as a negative family history does not exclude the diagnosis. Genetic counselors play an important role in patient support, education, and coordination with healthcare teams and advocacy organizations.
Nurses and allied health team members provide essential support through patient counseling, care coordination, and reinforcement of management strategies. Physical and occupational therapists individualize rehabilitation programs to improve strength, proprioception, joint stability, and functional independence. Interventions may include therapeutic exercise, bracing, assistive devices, and ergonomic modifications.[41] Dentists also play a role in early recognition and management, particularly in patients with oral and craniofacial manifestations.[71] Given the chronic nature of EDS and the absence of curative therapy, psychosocial support is a key component of care. Patients often face significant physical limitations, chronic pain, and activity-related challenges. Mental health professionals can help with coping strategies, while clear communication among interprofessional team members helps ensure consistent messaging and improved patient outcomes.[9][20]
Media
(Click Image to Enlarge)
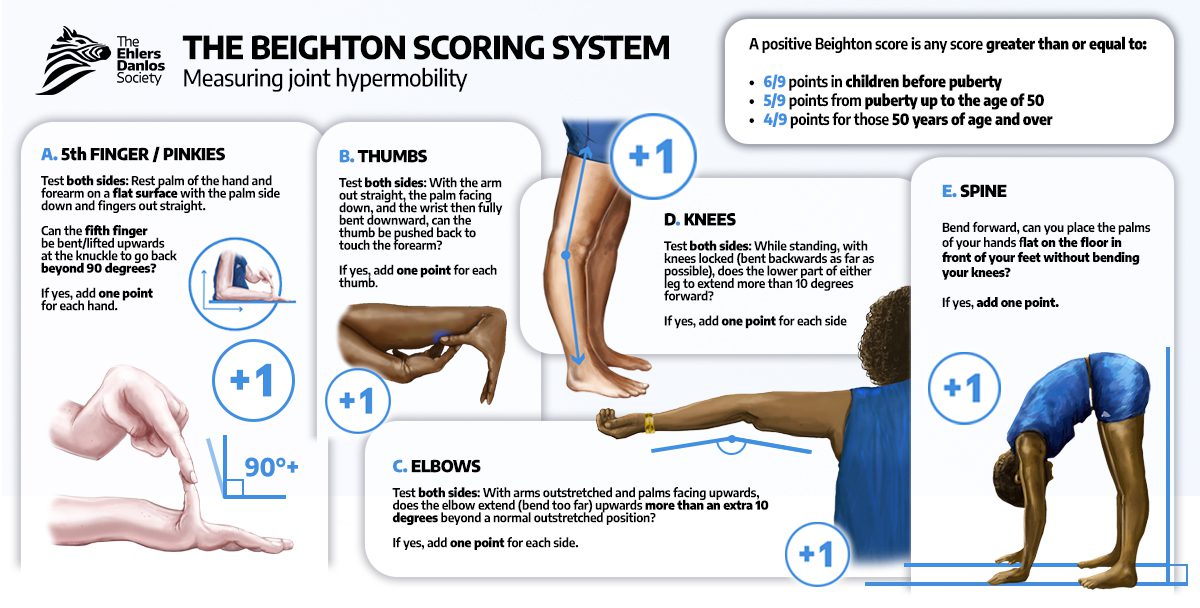
Beighton Scoring System. The Beighton Scoring System is a validated 9-point clinical assessment tool designed to quantify generalized joint hypermobility. It provides a standardized metric to assist in the screening of connective tissue disorders, such as hEDS and HSD, by evaluating five specific anatomical sites—the fifth metacarpophalangeal joints, the thumbs, elbows, knees, and the lumbar spine.
With permission of The Ehlers-Danlos Society
(Click Image to Enlarge)
References
Cortini F, Villa C. Ehlers-Danlos syndromes and epilepsy: An updated review. Seizure. 2018 Apr:57():1-4. doi: 10.1016/j.seizure.2018.02.013. Epub 2018 Mar 2 [PubMed PMID: 29499446]
Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B. The 2017 international classification of the Ehlers-Danlos syndromes. American journal of medical genetics. Part C, Seminars in medical genetics. 2017 Mar:175(1):8-26. doi: 10.1002/ajmg.c.31552. Epub [PubMed PMID: 28306229]
Castori M, Hakim A. Contemporary approach to joint hypermobility and related disorders. Current opinion in pediatrics. 2017 Dec:29(6):640-649. doi: 10.1097/MOP.0000000000000541. Epub [PubMed PMID: 28906340]
Level 3 (low-level) evidenceJunkiert-Czarnecka A, Pilarska-Deltow M, Bąk A, Heise M, Haus O. New variants in COL5A1 gene among Polish patients with Ehlers-Danlos syndrome: analysis of nine cases. Postepy dermatologii i alergologii. 2019 Feb:36(1):29-33. doi: 10.5114/ada.2018.79440. Epub 2019 Feb 22 [PubMed PMID: 30858776]
Level 3 (low-level) evidenceMalfait F, Castori M, Francomano CA, Giunta C, Kosho T, Byers PH. The Ehlers-Danlos syndromes. Nature reviews. Disease primers. 2020 Jul 30:6(1):64. doi: 10.1038/s41572-020-0194-9. Epub 2020 Jul 30 [PubMed PMID: 32732924]
Doolan BJ, Lavallee ME, Hausser I, Schubart JR, Michael Pope F, Seneviratne SL, Winship IM, Burrows NP. Extracutaneous features and complications of the Ehlers-Danlos syndromes: A systematic review. Frontiers in medicine. 2023:10():1053466. doi: 10.3389/fmed.2023.1053466. Epub 2023 Jan 23 [PubMed PMID: 36756177]
Level 1 (high-level) evidenceByers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, Johnson D, Pepin M, Robert L, Sanders L, Wheeldon N. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. American journal of medical genetics. Part C, Seminars in medical genetics. 2017 Mar:175(1):40-47. doi: 10.1002/ajmg.c.31553. Epub [PubMed PMID: 28306228]
Colman M, Syx D, De Wandele I, Dhooge T, Symoens S, Malfait F. Clinical and molecular characteristics of 168 probands and 65 relatives with a clinical presentation of classical Ehlers-Danlos syndrome. Human mutation. 2021 Oct:42(10):1294-1306. doi: 10.1002/humu.24258. Epub 2021 Jul 26 [PubMed PMID: 34265140]
Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genetics in medicine : official journal of the American College of Medical Genetics. 2010 Oct:12(10):597-605. doi: 10.1097/GIM.0b013e3181eed412. Epub [PubMed PMID: 20847697]
Micale L, Fusco C, Castori M. Ehlers-Danlos Syndromes, Joint Hypermobility and Hypermobility Spectrum Disorders. Advances in experimental medicine and biology. 2021:1348():207-233. doi: 10.1007/978-3-030-80614-9_9. Epub [PubMed PMID: 34807421]
Level 3 (low-level) evidenceLegrand A, Devriese M, Dupuis-Girod S, Simian C, Venisse A, Mazzella JM, Auribault K, Adham S, Frank M, Albuisson J, Jeunemaitre X. Frequency of de novo variants and parental mosaicism in vascular Ehlers-Danlos syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2019 Jul:21(7):1568-1575. doi: 10.1038/s41436-018-0356-2. Epub 2018 Nov 26 [PubMed PMID: 30474650]
Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, Giunta C, Kapferer-Seebacher I, Kosho T, Mendoza-Londono R, Pope MF, Rohrbach M, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Zschocke J, Malfait F. The Ehlers-Danlos syndromes, rare types. American journal of medical genetics. Part C, Seminars in medical genetics. 2017 Mar:175(1):70-115. doi: 10.1002/ajmg.c.31550. Epub [PubMed PMID: 28306225]
Hakim A, Grahame R. Joint hypermobility. Best practice & research. Clinical rheumatology. 2003 Dec:17(6):989-1004 [PubMed PMID: 15123047]
Kulas Søborg ML, Leganger J, Quitzau Mortensen L, Rosenberg J, Burcharth J. Establishment and baseline characteristics of a nationwide Danish cohort of patients with Ehlers-Danlos syndrome. Rheumatology (Oxford, England). 2017 May 1:56(5):763-767. doi: 10.1093/rheumatology/kew478. Epub [PubMed PMID: 28077691]
Demmler JC, Atkinson MD, Reinhold EJ, Choy E, Lyons RA, Brophy ST. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: a national electronic cohort study and case-control comparison. BMJ open. 2019 Nov 4:9(11):e031365. doi: 10.1136/bmjopen-2019-031365. Epub 2019 Nov 4 [PubMed PMID: 31685485]
Level 2 (mid-level) evidenceCarpenter SL, Abshire TC, Killough E, Anderst JD, AAP SECTION ON HEMATOLOGY/ONCOLOGY, THE AMERICAN SOCIETY OF PEDIATRIC HEMATOLOGY AND ONCOLOGY, and the AAP COUNCIL ON CHILD ABUSE AND NEGLECT. Evaluating for Suspected Child Abuse: Conditions That Predispose to Bleeding. Pediatrics. 2022 Oct 1:150(4):. pii: e2022059277. doi: 10.1542/peds.2022-059277. Epub [PubMed PMID: 36120799]
Lavanya K, Mahtani K, Abbott J, Jain A, Selvam P, Atwal H, Farres H, Atwal PS. A patient with a novel pathogenic variant in COL5A1 exhibiting prominent vascular and cardiac features. American journal of medical genetics. Part A. 2022 Jul:188(7):2192-2197. doi: 10.1002/ajmg.a.62745. Epub 2022 Apr 9 [PubMed PMID: 35396906]
Gensemer C, Burks R, Kautz S, Judge DP, Lavallee M, Norris RA. Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes. Developmental dynamics : an official publication of the American Association of Anatomists. 2021 Mar:250(3):318-344. doi: 10.1002/dvdy.220. Epub 2020 Aug 17 [PubMed PMID: 32629534]
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). American journal of medical genetics. 1998 Apr 28:77(1):31-7 [PubMed PMID: 9557891]
Level 1 (high-level) evidenceCallewaert B, Malfait F, Loeys B, De Paepe A. Ehlers-Danlos syndromes and Marfan syndrome. Best practice & research. Clinical rheumatology. 2008 Mar:22(1):165-89. doi: 10.1016/j.berh.2007.12.005. Epub [PubMed PMID: 18328988]
Syx D, Malfait F. Pathogenic mechanisms in genetically defined Ehlers-Danlos syndromes. Trends in molecular medicine. 2024 Sep:30(9):824-843. doi: 10.1016/j.molmed.2024.06.001. Epub 2024 Aug 14 [PubMed PMID: 39147618]
Chiarelli N, Ritelli M, Zoppi N, Colombi M. Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers‒Danlos Syndromes. Genes. 2019 Aug 12:10(8):. doi: 10.3390/genes10080609. Epub 2019 Aug 12 [PubMed PMID: 31409039]
De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clinical genetics. 2012 Jul:82(1):1-11. doi: 10.1111/j.1399-0004.2012.01858.x. Epub 2012 Mar 15 [PubMed PMID: 22353005]
Yew KS, Kamps-Schmitt KA, Borge R. Hypermobile Ehlers-Danlos Syndrome and Hypermobility Spectrum Disorders. American family physician. 2021 Apr 15:103(8):481-492 [PubMed PMID: 33856167]
Scheper MC, Nicholson LL, Adams RD, Tofts L, Pacey V. The natural history of children with joint hypermobility syndrome and Ehlers-Danlos hypermobility type: a longitudinal cohort study. Rheumatology (Oxford, England). 2017 Dec 1:56(12):2073-2083. doi: 10.1093/rheumatology/kex148. Epub [PubMed PMID: 28431150]
Yassin OM, Rihani FB. Multiple developmental dental anomalies and hypermobility type Ehlers-Danlos syndrome. The Journal of clinical pediatric dentistry. 2006 Summer:30(4):337-41 [PubMed PMID: 16937863]
Level 3 (low-level) evidenceKapferer-Seebacher I, Oakley-Hannibal E, Lepperdinger U, Johnson D, Ghali N, Brady AF, Sobey G, Zschocke J, van Dijk FS. Prospective clinical investigations of children with periodontal Ehlers-Danlos syndrome identify generalized lack of attached gingiva as a pathognomonic feature. Genetics in medicine : official journal of the American College of Medical Genetics. 2021 Feb:23(2):316-322. doi: 10.1038/s41436-020-00985-y. Epub 2020 Oct 2 [PubMed PMID: 33005042]
Ritelli M, Venturini M, Cinquina V, Chiarelli N, Colombi M. Multisystemic manifestations in a cohort of 75 classical Ehlers-Danlos syndrome patients: natural history and nosological perspectives. Orphanet journal of rare diseases. 2020 Jul 31:15(1):197. doi: 10.1186/s13023-020-01470-0. Epub 2020 Jul 31 [PubMed PMID: 32736638]
Level 3 (low-level) evidenceTofts LJ, Simmonds J, Schwartz SB, Richheimer RM, O'Connor C, Elias E, Engelbert R, Cleary K, Tinkle BT, Kline AD, Hakim AJ, van Rossum MAJ, Pacey V. Pediatric joint hypermobility: a diagnostic framework and narrative review. Orphanet journal of rare diseases. 2023 May 4:18(1):104. doi: 10.1186/s13023-023-02717-2. Epub 2023 May 4 [PubMed PMID: 37143135]
Level 3 (low-level) evidenceDamseh N, Dupuis L, O'Connor C, Oh RY, Wang YW, Stavropoulos DJ, Schwartz SB, Mendoza-Londono R. Diagnostic outcomes for molecular genetic testing in children with suspected Ehlers-Danlos syndrome. American journal of medical genetics. Part A. 2022 May:188(5):1376-1383. doi: 10.1002/ajmg.a.62672. Epub 2022 Feb 6 [PubMed PMID: 35128800]
Forghani I, See J, McGonigle WC. Hypermobile Ehlers-Danlos Syndrome: Diagnostic Challenges and the Role of Genetic Testing. Genes. 2025 Apr 29:16(5):. doi: 10.3390/genes16050530. Epub 2025 Apr 29 [PubMed PMID: 40428350]
Henneton P, Albuisson J, Adham S, Legrand A, Mazzella JM, Jeunemaitre X, Frank M. Accuracy of Clinical Diagnostic Criteria for Patients With Vascular Ehlers-Danlos Syndrome in a Tertiary Referral Centre. Circulation. Genomic and precision medicine. 2019 Mar:12(3):e001996. doi: 10.1161/CIRCGEN.117.001996. Epub [PubMed PMID: 30919682]
Hamonet C, Frédy D, Lefèvre JH, Bourgeois-Gironde S, Zeitoun JD. Brain injury unmasking Ehlers-Danlos syndromes after trauma: the fiber print. Orphanet journal of rare diseases. 2016 Apr 22:11():45. doi: 10.1186/s13023-016-0428-9. Epub 2016 Apr 22 [PubMed PMID: 27102338]
Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. The Journal of pediatrics. 2011 May:158(5):826-830.e1. doi: 10.1016/j.jpeds.2010.11.023. Epub 2010 Dec 28 [PubMed PMID: 21193204]
Level 2 (mid-level) evidenceUmekoji A, Fukai K, Hosomi N, Ishii M, Tanaka A, Murakami K, Kamoi H, Mizoguchi M, Utani A. Vascular type of Ehlers-Danlos syndrome associated with mild haemophilia A. Clinical and experimental dermatology. 2009 Jan:34(1):101. doi: 10.1111/j.1365-2230.2008.02794.x. Epub [PubMed PMID: 18828845]
Level 3 (low-level) evidenceSobey G. Ehlers-Danlos syndrome: how to diagnose and when to perform genetic tests. Archives of disease in childhood. 2015 Jan:100(1):57-61. doi: 10.1136/archdischild-2013-304822. Epub 2014 Jul 3 [PubMed PMID: 24994860]
Wallman D, Weinberg J, Hohler AD. Ehlers-Danlos Syndrome and Postural Tachycardia Syndrome: a relationship study. Journal of the neurological sciences. 2014 May 15:340(1-2):99-102. doi: 10.1016/j.jns.2014.03.002. Epub 2014 Mar 11 [PubMed PMID: 24685354]
Level 2 (mid-level) evidenceMathias CJ, Low DA, Iodice V, Owens AP, Kirbis M, Grahame R. Postural tachycardia syndrome--current experience and concepts. Nature reviews. Neurology. 2011 Dec 6:8(1):22-34. doi: 10.1038/nrneurol.2011.187. Epub 2011 Dec 6 [PubMed PMID: 22143364]
Garreth Brittain M, Flanagan S, Foreman L, Teran-Wodzinski P. Physical therapy interventions in generalized hypermobility spectrum disorder and hypermobile Ehlers-Danlos syndrome: a scoping review. Disability and rehabilitation. 2024 May:46(10):1936-1953. doi: 10.1080/09638288.2023.2216028. Epub 2023 May 25 [PubMed PMID: 37231592]
Level 2 (mid-level) evidenceMinhas D. Practical management strategies for benign hypermobility syndromes. Current opinion in rheumatology. 2021 May 1:33(3):249-254. doi: 10.1097/BOR.0000000000000798. Epub [PubMed PMID: 33741806]
Level 3 (low-level) evidenceAdam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Hakim A. Hypermobile Ehlers-Danlos Syndrome. GeneReviews(®). 1993:(): [PubMed PMID: 20301456]
Whalen KC, Crone W. Multidisciplinary Approach to Treating Chronic Pain in Patients with Ehlers-Danlos Syndrome: Critically Appraised Topic. Journal of pain research. 2022:15():2893-2904. doi: 10.2147/JPR.S377790. Epub 2022 Sep 13 [PubMed PMID: 36124037]
Chetty SP, Shaffer BL, Norton ME. Management of pregnancy in women with genetic disorders, Part 1: Disorders of the connective tissue, muscle, vascular, and skeletal systems. Obstetrical & gynecological survey. 2011 Nov:66(11):699-709. doi: 10.1097/OGX.0b013e31823cdd50. Epub [PubMed PMID: 22186601]
Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Malfait F, Symoens S, Syx D. Classic Ehlers-Danlos Syndrome. GeneReviews(®). 1993:(): [PubMed PMID: 20301422]
Saputra PBT, Widiarti W, Siahaan PP, Putra RM, Putranto JNE, Budiarto RM, Luthfah N, Multazam CECZ, D'Oria M, Alkaff FF. The Impact of Celiprolol in Vascular Ehlers-Danlos Syndrome: A Systematic Review of Current Evidence. Medical sciences (Basel, Switzerland). 2025 Jun 9:13(2):. doi: 10.3390/medsci13020074. Epub 2025 Jun 9 [PubMed PMID: 40559232]
Level 1 (high-level) evidenceMathias CJ, Owens A, Iodice V, Hakim A. Dysautonomia in the Ehlers-Danlos syndromes and hypermobility spectrum disorders-With a focus on the postural tachycardia syndrome. American journal of medical genetics. Part C, Seminars in medical genetics. 2021 Dec:187(4):510-519. doi: 10.1002/ajmg.c.31951. Epub 2021 Nov 12 [PubMed PMID: 34766441]
Lian T, Bhandari A, Shalhub S. What Every Vascular Surgeon Should Know About Vascular Ehlers-Danlos Syndrome. Annals of vascular surgery. 2026 Aug:129():302-306. doi: 10.1016/j.avsg.2026.03.032. Epub 2026 Mar 27 [PubMed PMID: 41905459]
Yonko EA, LoTurco HM, Carter EM, Raggio CL. Orthopedic considerations and surgical outcomes in Ehlers-Danlos syndromes. American journal of medical genetics. Part C, Seminars in medical genetics. 2021 Dec:187(4):458-465. doi: 10.1002/ajmg.c.31958. Epub 2021 Nov 29 [PubMed PMID: 34845816]
Kang J, Hanif M, Mirza E, Jaleel S. Ehlers-Danlos Syndrome in Pregnancy: A Review. European journal of obstetrics, gynecology, and reproductive biology. 2020 Dec:255():118-123. doi: 10.1016/j.ejogrb.2020.10.033. Epub 2020 Oct 17 [PubMed PMID: 33113401]
Zoppi N, Chiarelli N, Binetti S, Ritelli M, Colombi M. Dermal fibroblast-to-myofibroblast transition sustained by αvß3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochimica et biophysica acta. Molecular basis of disease. 2018 Apr:1864(4 Pt A):1010-1023. doi: 10.1016/j.bbadis.2018.01.005. Epub 2018 Jan 5 [PubMed PMID: 29309923]
Colombi M, Dordoni C, Chiarelli N, Ritelli M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/ehlers-danlos syndrome hypermobility type compared to other heritable connective tissue disorders. American journal of medical genetics. Part C, Seminars in medical genetics. 2015 Mar:169C(1):6-22. doi: 10.1002/ajmg.c.31429. Epub [PubMed PMID: 25821090]
Van Laer L, Dietz H, Loeys B. Loeys-Dietz syndrome. Advances in experimental medicine and biology. 2014:802():95-105. doi: 10.1007/978-94-007-7893-1_7. Epub [PubMed PMID: 24443023]
Level 3 (low-level) evidenceBerk DR, Bentley DD, Bayliss SJ, Lind A, Urban Z. Cutis laxa: a review. Journal of the American Academy of Dermatology. 2012 May:66(5):842.e1-17. doi: 10.1016/j.jaad.2011.01.004. Epub 2012 Mar 2 [PubMed PMID: 22387031]
Berglund B, Nordström G, Lützén K. Living a restricted life with Ehlers-Danlos syndrome (EDS). International journal of nursing studies. 2000 Apr:37(2):111-8 [PubMed PMID: 10684952]
Owen SM, Durst RD. Ehlers-Danlos syndrome simulating child abuse. Archives of dermatology. 1984 Jan:120(1):97-101 [PubMed PMID: 6691721]
Level 3 (low-level) evidenceCollins Hutchinson ML, Liang E, Fuster E, Blitshteyn S. Autonomic symptom burden, comorbidities and quality of life in women with Hypermobility Spectrum Disorders and hypermobile Ehlers-Danlos syndrome. Autonomic neuroscience : basic & clinical. 2025 Dec:262():103356. doi: 10.1016/j.autneu.2025.103356. Epub 2025 Oct 14 [PubMed PMID: 41118678]
Level 2 (mid-level) evidenceKalisch L, Hamonet C, Bourdon C, Montalescot L, de Cazotte C, Baeza-Velasco C. Predictors of pain and mobility disability in the hypermobile Ehlers-Danlos syndrome. Disability and rehabilitation. 2020 Dec:42(25):3679-3686. doi: 10.1080/09638288.2019.1608595. Epub 2019 May 7 [PubMed PMID: 31060411]
Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. The New England journal of medicine. 2000 Mar 9:342(10):673-80 [PubMed PMID: 10706896]
Lum YW, Brooke BS, Black JH 3rd. Contemporary management of vascular Ehlers-Danlos syndrome. Current opinion in cardiology. 2011 Nov:26(6):494-501. doi: 10.1097/HCO.0b013e32834ad55a. Epub [PubMed PMID: 21852761]
Level 3 (low-level) evidenceFrank M, Adham S, Seigle S, Legrand A, Mirault T, Henneton P, Albuisson J, Denarié N, Mazzella JM, Mousseaux E, Messas E, Boutouyrie P, Jeunemaitre X. Vascular Ehlers-Danlos Syndrome: Long-Term Observational Study. Journal of the American College of Cardiology. 2019 Apr 23:73(15):1948-1957. doi: 10.1016/j.jacc.2019.01.058. Epub [PubMed PMID: 30999998]
Level 2 (mid-level) evidenceWenstrup RJ, Meyer RA, Lyle JS, Hoechstetter L, Rose PS, Levy HP, Francomano CA. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2002 May-Jun:4(3):112-7 [PubMed PMID: 12180144]
Level 2 (mid-level) evidenceAwad AA, Yappalparvi A, Khatib MN, R R, Kaur M, Srivastava M, Barwal A, Prasad GVS, Rajput P, Syed R, Sharma G, Prasoon A, Shabil M, Punia A, Jagga M, Mehta R, Sah S, Satapathy P, Gaidhane AM, Mawejje E, Bushi G. Prevalence of vascular complications in Ehlers-Danlos syndrome: a systematic review and meta-analysis. Orphanet journal of rare diseases. 2025 Jun 20:20(1):312. doi: 10.1186/s13023-025-03854-6. Epub 2025 Jun 20 [PubMed PMID: 40542421]
Level 1 (high-level) evidenceMorris SA, Flyer JN, Yetman AT, Quezada E, Cappella ES, Dietz HC, Milewicz DM, Ouzounian M, Rigelsky CM, Tierney S, Lacro RV, American Heart Association Council on Lifelong Congenital Heart Disease and Heart Health in the Young (Young Hearts); Council on Cardiovascular and Stroke Nursing; Council on Peripheral Vascular Disease; Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; and Council on Cardiovascular Surgery and Anesthesia. Cardiovascular Management of Aortopathy in Children: A Scientific Statement From the American Heart Association. Circulation. 2024 Sep 10:150(11):e228-e254. doi: 10.1161/CIR.0000000000001265. Epub 2024 Aug 12 [PubMed PMID: 39129620]
Park MA, Shin SY, Kim YJ, Park MJ, Lee SH. Vascular Ehlers-Danlos syndrome with cryptorchidism, recurrent pneumothorax, and pulmonary capillary hemangiomatosis-like foci: A case report. Medicine. 2017 Nov:96(47):e8853. doi: 10.1097/MD.0000000000008853. Epub [PubMed PMID: 29381997]
Level 3 (low-level) evidenceShirley ED, Demaio M, Bodurtha J. Ehlers-danlos syndrome in orthopaedics: etiology, diagnosis, and treatment implications. Sports health. 2012 Sep:4(5):394-403 [PubMed PMID: 23016112]
Wright GL, Wen T, Engel DJ, Guglielminotti J, Andrikopoulou M, Booker WA, D'Alton ME, Friedman AM. Delivery Outcomes and Postpartum Readmissions Associated with Ehlers-Danlos Syndrome. American journal of perinatology. 2024 May:41(S 01):e3045-e3051. doi: 10.1055/a-2185-4149. Epub 2023 Oct 4 [PubMed PMID: 37793432]
Haem T, Benson B, Dernoncourt A, Gondry J, Schmidt J, Foulon A. Vascular Ehlers-Danlos syndrome and pregnancy: A systematic review. BJOG : an international journal of obstetrics and gynaecology. 2024 Nov:131(12):1620-1629. doi: 10.1111/1471-0528.17893. Epub 2024 Jun 26 [PubMed PMID: 38926786]
Level 1 (high-level) evidenceHurst BS, Lange SS, Kullstam SM, Usadi RS, Matthews ML, Marshburn PB, Templin MA, Merriam KS. Obstetric and gynecologic challenges in women with Ehlers-Danlos syndrome. Obstetrics and gynecology. 2014 Mar:123(3):506-513. doi: 10.1097/AOG.0000000000000123. Epub [PubMed PMID: 24499752]
Guilleminault C, Primeau M, Chiu HY, Yuen KM, Leger D, Metlaine A. Sleep-disordered breathing in Ehlers-Danlos syndrome: a genetic model of OSA. Chest. 2013 Nov:144(5):1503-1511. doi: 10.1378/chest.13-0174. Epub [PubMed PMID: 23929538]
Level 2 (mid-level) evidenceHarris J, Bartlett M, Baker D, Berlin C, Bowen J, Cummings C, Fallows C, Green C, Griffin J, Julier K, Kammin T, Sehra R, Stacey C, Cobben J, Ghali N, Johnson D, Sobey G, van Dijk FS. An exemplary model of genetic counselling for highly specialised services. Journal of community genetics. 2023 Apr:14(2):115-119. doi: 10.1007/s12687-023-00640-4. Epub 2023 Mar 9 [PubMed PMID: 36892793]
Cortés-Bretón Brinkmann J, García-Gil I, Lobato-Peña DM, Martínez-Mera C, Suárez-García MJ, Martínez-González JM, Rioboo M. The key role of the dental practitioner in early diagnosis of periodontal Ehlers-Danlos syndromes: a rare case report of siblings. Quintessence international (Berlin, Germany : 1985). 2021:52(2):166-174. doi: 10.3290/j.qi.a45263. Epub [PubMed PMID: 33433082]
Level 3 (low-level) evidence