Introduction
Down syndrome is the most common viable autosomal trisomy, resulting from an extra copy of chromosome 21, and is the leading genetic cause of intellectual disability.[1][2][3]
Down syndrome was first described in 1866 by the English physician John Langdon Down, who recognized its distinct constellation of physical and developmental features. The genetic basis of this condition was established in 1959 at the Paris laboratory of pediatrician Raymond Turpin, where trisomy 21 was identified by his students, Marthe Gautier and Jérôme Lejeune. The attribution of this discovery remains debated, particularly concerning the respective contributions of Gautier and Lejeune.[4]
Down syndrome, also known as trisomy 21, is characterized by a recognizable pattern of congenital anomalies and neurodevelopmental differences, with variable involvement of multiple organ systems. Although life expectancy has improved significantly in recent decades, individuals with Down syndrome experience unique health needs throughout their lifespan, including heightened risks of heart disease if congenital heart defects are present, hematologic disorders, autoimmune diseases, sleep-disordered breathing, sensory impairments, and neurological conditions such as early-onset Alzheimer disease. Updated guidance emphasizes the importance of family-centered counseling, early detection of comorbidities, and coordinated interprofessional care spanning childhood through adulthood (see Image. Newborn with Down Syndrome).[5][6][7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The majority of patients with Down syndrome have an extra copy of chromosome 21 (see Images. Karyotype of Down Syndrome [Trisomy 21]). Various hypotheses have been proposed to explain the genetic basis of Down syndrome and the association between specific genotypes and clinical phenotypes. One such hypothesis is gene dosage imbalance, in which the increased copy number of genes located on human chromosome 21 (Hsa21) leads to overexpression of chromosome 21 genes and subsequent disruption of developmental pathways.[8] This hypothesis also includes the possibility that various genes may be associated with distinct phenotypes of Down syndrome. Another popular hypothesis is amplified developmental instability, which suggests that the genetic imbalance caused by multiple trisomic genes has a greater impact on the expression and regulation of numerous other genes.[8]
The critical region hypothesis is another well-recognized explanation. Down syndrome critical regions (DSCRs) are specific segments of chromosome 21 associated with partial trisomy of the Has21 region. In particular, the DSCR at 21q21.22 is responsible for many clinical features of Down syndrome.[8][9] Comprehensive analyses indicate that a single critical region gene cannot account for all the phenotypic features associated with trisomy 21. Rather, it is more evident that multiple critical regions or genes contribute to this syndrome’s characteristics.[10]
Epidemiology
The incidence of Down syndrome increases with maternal age, and its occurrence varies in different populations (1 in 319 to 1 in 1000 live births).[11][12] Although the frequency of Down syndrome in embryos is quite high at conception, an estimated 50% to 75% of affected fetuses are lost through miscarriage before term. The occurrence of other autosomal trisomies is much more frequent than trisomy 21, but their postnatal survival rates are much lower than those of Down syndrome. This high survival rate among patients with trisomy 21 is believed to be due to the small number of genes on chromosome 21 (Hsa21), which is the smallest and least gene-dense autosome.[13]
A 2023 cohort study found that between 2011 and 2018, during which numerous states in the United States enacted 20-week abortion bans, the incidence of Down syndrome diagnosed at birth increased nationwide, with a substantially greater rise observed in states that implemented these bans. These findings highlight the importance of considering local laws and social norms when counseling families following a prenatal diagnosis of Down syndrome.[14]
Pathophysiology
Down syndrome results from an extra copy of chromosome 21. Approximately 95% of cases result from meiotic nondisjunction, in which chromosome 21 fails to separate during gametogenesis. About 3% to 4% of cases involve Robertsonian translocations, most commonly the fusion of the long arms of chromosomes 14 and 21 (der[14;21]), and 1% to 2% are mosaic, arising from postzygotic nondisjunction. In mosaicism, 2 distinct cell lines arise due to an error in cell division after fertilization.[11][15][16]
History and Physical
Clinical Features
Down syndrome affects multiple organ systems and is associated with a wide range of clinical conditions. Patients display a wide array of signs and symptoms, such as intellectual and developmental disabilities, neurological features, congenital heart defects, gastrointestinal abnormalities, characteristic facial features, and other abnormalities (see Image. Facial Features of a Child With Down Syndrome).[17]
Congenital Heart Defects
Congenital heart defects are by far the most common congenital malformations and the leading cause of morbidity and mortality in individuals with Down syndrome, particularly during the first 2 years of life. Although various explanations have been proposed for geographic and seasonal variation in the occurrence of different types of congenital cardiac defects observed in trisomy 21, the results have been inconclusive so far.[18]
Congenital heart defects occur in up to 50% of infants born with Down syndrome. The most prevalent anomaly is an atrioventricular septal defect (AVSD), also known as an atrioventricular canal or endocardial cushion defect, which accounts for approximately 40% of congenital heart defects in this population. This condition is associated with a mutation in the non-Hsa21 CRELD1 gene.[11][19] The second most common cardiac defect in Down syndrome is a ventricular septal defect (VSD), which is observed in about 32% of the patients with Down syndrome. Together with AVSD, these account for more than 50% of congenital cardiac malformations in patients with Down syndrome.[11][19][20]
Other cardiac defects associated with trisomy 21 include secundum atrial septal defect (ASD; 10%), tetralogy of Fallot (6%), and isolated patent ductus arteriosus (PDA; 4%); about 30% of patients have more than one cardiac malformation. The prevalence of specific defects varies geographically, with VSD being the most common in Asia and secundum-type ASD predominating in Latin America. The reasons for these regional differences in the prevalence of congenital heart defect subtypes remain unclear; however, contributing factors may include geographic proximity between populations, shared genetic and environmental influences, and similarities in healthcare practices.[11]
Given the high prevalence of congenital heart defects in individuals with Down syndrome, it is recommended that all patients undergo an echocardiogram within the first few weeks of life.
Gastrointestinal Tract Manifestations
Individuals with trisomy 21 can have many structural and functional disorders related to the gastrointestinal tract. Structural defects may occur anywhere from the mouth to the anus, with specific defects, such as duodenal and small bowel atresia or stenosis, annular pancreas, imperforate anus, and Hirschsprung disease, being more common in this population than in the general population.[2]
About 2% of patients with Down syndrome have Hirschsprung disease, while 12% of patients with Hirschsprung disease have Down syndrome.[2][11] Hirschsprung disease is a form of functional lower intestinal obstruction in which neural cells fail to migrate into the distal segment of the rectum, leaving an aganglionic segment without normal peristalsis. This disrupts the normal defecation reflex, leading to a functional obstruction.[21] Affected infants usually present with signs and symptoms related to intestinal obstruction. Duodenal atresia and imperforate anus typically present during the neonatal period.[22]
Down syndrome patients are also prone to other gastrointestinal disorders such as gastroesophageal reflux (GERD), chronic constipation, intermittent diarrhea, and celiac disease.[23] Because celiac disease occurs in approximately 5% of individuals with Down syndrome, annual screening is recommended. Patients diagnosed with celiac disease must adhere to a lifelong gluten-free diet.
Hematological Disorders
Several hematological disorders are associated with Down syndrome. The hematological abnormalities in a newborn with Down syndrome (HANDS) constitute neutrophilia, thrombocytopenia, and polycythemia, which are observed in 80%, 66%, and 34% of infants, respectively. HANDS is usually mild and resolves within the first 3 weeks of life.[24][25][26]
Another disorder specific to Down syndrome is a transient myeloproliferative disorder, which is defined as the presence of blast cells in infants younger than 3 months. This condition is characterized by clonal proliferation of megakaryocytes, detected during the first week of life, and resolves by 3 months of age. This condition is also known as transient abnormal myelopoiesis or transient leukemia and is known to be present in about 10% of patients with Down syndrome. If this condition occurs in the fetus, it can lead to spontaneous abortion.[27][28]
Individuals with Down syndrome are 10 times more likely to develop leukemia than the general pediatric population [29] and constitute about 2% of all pediatric acute lymphoblastic leukemia (ALL) cases and 10% of all pediatric acute myeloid leukemia (AML) cases. Children with Down syndrome aged 5 or younger have an elevated risk of AML, particularly acute megakaryoblastic leukemia. This increased risk is believed to be related to alterations in hematopoiesis associated with trisomy 21 and an increased susceptibility of myeloid progenitor cells to leukemogenic mutations. Although ALL also occurs at increased rates, its incidence rises later in childhood and does not exceed AML in children aged 5 or younger.[30] Approximately 30% of individuals with Down syndrome who develop ALL harbor a functional mutation in the Janus kinase 2 (JAK2) gene.[31][32]
Approximately 10% of neonates with Down syndrome develop transient myeloproliferative disorder (TMD), and 20% to 30% of those affected subsequently develop acute megakaryoblastic leukemia (AMKL) before 4 years of age. AMKL is associated with the GATA1 gene, an X-linked transcription factor that leads to abnormal proliferation of immature megakaryocytes.[33]
Neurological Disorders
Trisomy of Hsa21 is associated with reduced brain volume, particularly in the hippocampus and cerebellum.[34] Hypotonia is a hallmark feature of infants with Down syndrome, occurring in nearly all cases. The condition is defined as decreased resistance to passive muscle stretch and is responsible for delayed motor development in these patients.[35]. Because of hypotonia, patients exhibit joint laxity, which decreases gait stability and increases the energy required for physical exertion.[36]. Individuals with Down syndrome are prone to reduced bone mass and an increased risk of fractures due to reduced physical activity,[34][37] while ligamentous laxity predisposes these patients to atlantoaxial subluxation.[38]
Approximately 5% to 13% of children with Down syndrome experience seizures.[39] Among these, 40% have onset before their first year of life, with infantile spasms being the most common seizure type in this age group.[40] Children with Down syndrome and infantile spasms tend to respond more favorably to antiepileptic therapy than other children with the same condition. Early recognition and treatment are therefore critical, as timely intervention can improve developmental outcomes.[39]
Lennox-Gastaut syndrome is also more prevalent in children with Down syndrome. When it occurs, it typically has a late onset and is associated with reflex seizures, along with an increased rate of electroencephalography (EEG) abnormalities.[41]
Approximately 40% of individuals with Down syndrome develop tonic-clonic or myoclonic seizures within their first 3 decades.[40] Dementia occurs more commonly in patients aged 45 or older,[42] and approximately 84% of patients are prone to developing seizures.[43] Seizures appear to be associated with the rapid decline in cognitive function.[44]
The risk of developing early-onset Alzheimer disease is high in patients with Down syndrome, with 50% to 70% developing dementia by age 60.[45] Amyloid precursor protein (APP), which is encoded on Hsa21, is known to be associated with an increased risk of Alzheimer disease, and trisomy of this protein is likely to contribute to the increased frequency of dementia in this population. Recent studies have also shown that triplication of APP is associated with an increased risk of early-onset Alzheimer disease in the general population.[46][37]
Nearly all patients with Down syndrome have mild to moderate intellectual and learning disabilities. Trisomy of multiple genes, including DYRK1A, synaptojanin 1, and single-minded homolog 2 (SIM2), has been shown to cause learning and memory deficits in mice, suggesting that overexpression of these genes may cause cognitive disabilities in individuals with Down syndrome.[47]
Endocrinological Disorders
Thyroid dysfunction is common in individuals with Down syndrome. Hypothyroidism may be present at birth (congenital) or develop later in life (acquired).[48] The newborn screening program in New York has reported an increased incidence of congenital hypothyroidism in infants with Down syndrome compared with the general population.[49] Anti-thyroid autoantibodies were detected in 13% to 34% of patients with Down syndrome who developed acquired hypothyroidism, and their concentrations increased after age 8.[48] About half of individuals with Down syndrome exhibit subclinical hypothyroidism, with elevated thyroid-stimulating hormone (TSH) and normal thyroxine levels.[50] Hyperthyroidism is far less common than hypothyroidism, although its prevalence is higher than that in the general pediatric population.[51]
Abnormalities in sexual development are also common, with delayed puberty observed in both boys and girls. In girls, primary hypogonadism presents as delayed menarche or adrenarche, while in boys it can manifest as cryptorchidism, ambiguous genitalia, micropenis, small testes, low sperm count, and scanty growth of axillary and pubic hair.[48]
A deficiency in insulin-like growth factor (IGF) is believed to contribute to delayed skeletal maturation and short stature in individuals with Down syndrome. Alterations in the IGF pathway underlie the skeletal phenotype, characterized by short stature, delayed bone development, reduced bone mineral density, and an increased risk of early-onset osteoporosis.[48][52]
Musculoskeletal Disorders
Children with Down syndrome are at increased risk of reduced muscle mass due to hypotonia and ligamentous laxity, which can delay gross motor development and, in some cases, contribute to joint instability or dislocation.[53] Individuals with Down syndrome are also prone to vitamin D deficiency due to limited sunlight exposure, inadequate dietary intake, malabsorption associated with celiac disease, and increased metabolism from anticonvulsant therapy. These deficiencies contribute to reduced bone mass and an increased risk of recurrent fractures.[54]
Ophthalmologic Manifestations
Ocular and orbital anomalies are common in children with Down syndrome. These include blepharitis (2%-7%), keratoconus (5%-8%), cataracts (25%-85%), retinal anomalies (0%-38%), strabismus (23%-44%), amblyopia (10%- to 26%), nystagmus (5% to 30%), refractive errors (18%-58%), glaucoma (<1%), iris anomalies (38%-90%), and rarely, optic nerve anomalies (see Image. Eyes of an Infant With Down Syndrome).
Untreated ocular anomalies can significantly affect the quality of life for individuals with Down syndrome, who should have a comprehensive eye examination in the first 6 months of life and annually thereafter.[55]
Otorhinolaryngological Disorders
Ear, nose, and throat (ENT) disorders are common in individuals with Down syndrome. Anatomical differences in the ear predispose them to hearing deficits. Hearing loss is most often conductive, resulting from impacted cerumen and middle ear pathologies, including chronic middle ear effusion due to small Eustachian tubes, acute otitis media, and tympanic membrane perforation. Many affected children require surgical insertion of pressure-equalization tubes.
Sensorineural hearing loss is also associated with Down syndrome due to structural abnormalities in the inner ear, such as narrow internal auditory canals.[56]
Evaluation
Several methods are available for the prenatal diagnosis of Down syndrome. Ultrasound performed between 11 and 24 weeks of gestation can identify soft markers such as increased nuchal fold thickness, a small or absent nasal bone, and large ventricles.[57] Nuchal translucency, measured by ultrasound between 11 and 14 weeks of gestation, represents a collection of fluid beneath the skin at the posterior fetal neck. In fetuses with Down syndrome, increased nuchal translucency (greater than 3 mm) reflects abnormal lymphatic development and impaired fluid drainage, leading to fluid accumulation in the nuchal region during the first trimester. When nuchal translucency alone is used to modify the age-related risk of trisomy 21, the detection rate is approximately 70%.
Increased nuchal translucency may also be observed in other chromosomal abnormalities, including trisomy 13 (Patau syndrome), trisomy 18 (Edwards syndrome), and Turner syndrome. Amniocentesis and chorionic villus sampling have been widely used for definitive diagnosis, but there is a small risk of miscarriage, estimated at 0.5% to 1%.[58][59]
Several other methods have also been developed and used for the rapid prenatal and postnatal detection of trisomy 21. Fluorescence in situ hybridization (FISH) of interphase nuclei is most commonly performed using either Hsa21-specific probes or the entire Hsa21.[60] Another method currently used is quantitative fluorescent polymerase chain reaction (QF-PCR), which detects 3 different alleles using DNA polymorphic markers.[61] The accuracy of this technique depends on the availability of informative markers and adequate DNA samples. Studies have shown that up to 86.67% of Down syndrome cases can be identified using short tandem repeat (STR) marker analysis.[62]
A relatively new method, paralogous sequence quantification (PSQ), uses the paralogous sequence on the Hsa21 copy number. This technique is a PCR-based method that uses paralogous genes to detect targeted chromosome-number abnormalities, known as PSQ.[63]
Additional noninvasive prenatal diagnostic methods are under investigation for the prenatal detection of Down syndrome. These approaches rely on identifying fetal cells circulating in maternal blood or detecting cell-free fetal DNA in maternal serum. These methods aim to reduce the need for invasive procedures, such as amniocentesis, while maintaining high diagnostic accuracy.[64]
Cell-free fetal DNA accounts for approximately 5% to 10% of maternal plasma and increases during pregnancy, then clears after delivery. Although this method has been used to determine fetal Rh status in Rh-negative women,[65] fetal sex in sex-linked disorders,[66] and paternally inherited autosomal recessive and dominant traits, its use in identifying chromosomal aneuploidy, especially trisomy, remains challenging.[67][68]
Additional emerging techniques, including digital PCR and next-generation sequencing, are also being developed for the prenatal diagnosis of Down syndrome.[69]
Treatment / Management
Management of Down syndrome requires a multidisciplinary approach. Newborns suspected of having the condition should undergo karyotype testing to confirm the diagnosis. Families should be referred to a clinical geneticist for confirmatory genetic testing and comprehensive genetic counseling for both parents.
Education is a central component of managing Down syndrome. Clinicians should counsel parents and caregivers about the broad range of medical, developmental, and behavioral conditions associated with trisomy 21 to support early recognition, timely evaluation, and appropriate intervention. Management is primarily supportive and symptom-directed, with the overarching goals of optimizing health, development, functional capacity, and quality of life rather than achieving a cure.
Primary care clinicians play a critical role in the longitudinal treatment of individuals with Down syndrome. They serve as the central point of coordination for multidisciplinary care, ensuring appropriate screening, surveillance, referrals, and follow-up across the individual's lifespan. Ongoing continuity of care enables primary care providers to monitor the patient's growth and development, comorbid conditions, and family needs, while reinforcing anticipatory guidance and preventive care.
In 2022, the American Academy of Pediatrics (AAP) published updated health supervision guidelines for children and adolescents with Down syndrome. These guidelines provide age-specific, evidence-based recommendations from birth through early adulthood, including screening for congenital anomalies, endocrine disorders, vision and hearing impairments, developmental delays, and psychosocial needs. Adherence to these structured recommendations supports consistent, comprehensive care and helps reduce preventable morbidity in this population.[70]
In 2025, the American Academy of Family Physicians published complementary guidance offering practical implementation strategies for family physicians caring for children with Down syndrome. This guidance emphasizes the role of the patient-centered medical home in coordinating comprehensive care across multiple subspecialties.[71]
Core Principles
Key principles of care for individuals with Down syndrome include ongoing assessment at every health supervision visit; personal support for the family; participation in a family-centered medical home; screening and management of age-specific medical and developmental conditions related to Down syndrome; access to financial and medical support programs; injury and abuse prevention; and guidance on nutrition and activity to maintain a healthy weight.
Growth Monitoring
Growth monitoring in children with Down syndrome should utilize Down syndrome–specific growth charts for weight, length/height, and head circumference. Down syndrome–specific body mass index (BMI) charts are recommended for children aged 2 to 10. Beyond this age, transition to the Centers for Disease Control and Prevention (CDC) BMI-for-age charts is advised, as these better reflect excess adiposity in older children with Down syndrome.
The AAP emphasizes promoting healthy dietary patterns and active lifestyles at all ages to reduce the risk of obesity. Optimal growth and weight management require a balanced diet, regular physical activity, and, when indicated, physical therapy to support strength and motor development. Feeding difficulties and poor weight gain in infancy, particularly in children with congenital heart disease, often improve after corrective cardiac surgery.
Cardiac Evaluation
All newborns with Down syndrome should undergo echocardiography, regardless of prenatal screening or imaging results. Subsequent cardiology follow-up should be tailored to echocardiographic findings and the patient's clinical status. Infants with cardiac defects causing intracardiac left-to-right shunts should be monitored at every well-child visit for signs of congestive heart failure, including tachypnea, feeding difficulties, and poor weight gain, as pulmonary vascular resistance decreases.
Thyroid Screening
Thyroid evaluation includes reviewing newborn thyroid function screening results and measuring TSH at 6 and 12 months of age, followed by annual testing thereafter due to the increased risk of acquired thyroid disease. If antithyroid antibodies are detected, TSH should be measured every 6 months.
Hearing Evaluation
Hearing evaluation includes an ear-specific audiologic assessment within the first month of life, repeated at 6 months, followed by annual testing. If middle ear disease occurs, a developmentally appropriate hearing evaluation should be obtained after treatment. Structural ear anomalies place children at risk for both conductive and sensorineural hearing loss, as well as vestibular problems that affect balance.
Ophthalmologic Assessment
Ophthalmologic evaluation should be performed within the first 6 months of life to evaluate for strabismus, cataracts, nasolacrimal duct obstruction, refractive errors, glaucoma, and nystagmus. Thereafter, ongoing assessment is recommended either through photoscreening at every health supervision visit or by a pediatric ophthalmologist every 2 years.
Hematologic Monitoring
Hematological monitoring includes obtaining a complete blood count with differential and iron studies beginning at age 1 and annually thereafter to screen for iron deficiency. Clinicians should remain alert for signs and symptoms of leukemia and obtain additional bloodwork as indicated.
Atlantoaxial Instability Surveillance
Surveillance for atlantoaxial instability involves discussing cervical spine precautions with families at least every 2 years and performing a careful history and physical examination at each well-child visit, with attention to signs of myelopathy. Parents should be instructed about concerning symptoms, including changes in gait, bowel or bladder dysfunction, neck pain or stiffness, head tilt, or weakness. The AAP no longer recommends routine radiographic screening for atlantoaxial instability in asymptomatic children because neutral-position x-rays do not reliably predict who will develop symptomatic instability, and normal findings do not ensure future safety.
Sleep-Disordered Breathing
Proper breathing should be discussed during health supervision visits by inquiring about heavy breathing, snoring, unusual sleep positions, frequent nighttime awakenings, daytime sleepiness, apneic pauses, and behavioral problems.
Developmental Support
Developmental support includes reviewing early intervention services (such as physical, occupational, and speech therapy) at every health supervision visit. Families should be counseled on the transition from early intervention to preschool at 36 months, including the shift from an Individualized Family Service Plan to an Individualized Education Program. Screening for autism spectrum disorder between 18 and 24 months requires particular attention, as its presentation in children with Down syndrome may differ from typical autism spectrum disorder. Children with both conditions demonstrate better social relating, receptive language, and imitation skills than those with autism spectrum disorder alone. Careful use of standard, validated screening tools is essential to avoid missed diagnoses.
Additional Screening
Additional screening for individuals with Down syndrome includes evaluation for celiac disease in those with gastrointestinal symptoms, a neurologic examination at each visit, with consultation as needed for seizures or other neurologic dysfunction, and dermatologic assessment for common conditions such as xerosis and cutis marmorata. Testicular palpation should be performed at each health supervision visit starting in early childhood.
Anticipatory Guidance
For adolescents with Down syndrome, anticipatory guidance includes developmentally appropriate sexual health education, counseling on contraception, and discussion of the increased risk of sexual exploitation. Planning for the transition to adult healthcare is essential. Discussions with parents and caregivers should also address guardianship and long-term financial planning, promotion of self-care and personal hygiene skills, exploration of independent or supported living options, postsecondary education or vocational opportunities, and consideration of residential alternatives such as supervised apartments or group homes.
The comprehensive nature of these guidelines underscores the importance of coordinated, multidisciplinary care delivered through a family-centered medical home to effectively manage the complex medical, developmental, and psychosocial needs of children and adolescents with Down syndrome while supporting their families throughout childhood and adolescence. In addition to primary care, care teams often include developmental pediatricians, pediatric pulmonologists, gastroenterologists, neurologists, neurosurgeons, orthopedic specialists, child psychiatrists, physical and occupational therapists, speech-language therapists, and audiologists.
Differential Diagnosis
Several genetic and metabolic conditions may present with clinical features similar to those of Down syndrome and should be considered in the differential diagnosis. These include:
- Congenital hypothyroidism
- Mosaic trisomy 21 syndrome
- Partial trisomy 21 (or 21q duplication)
- Robertsonian trisomy 21
- Trisomy 18 and trisomy 13
- Zellweger syndrome and other peroxisomal disorders
- Single-gene or contiguous-gene syndromes associated with hypotonia and characteristic facial features (eg, Noonan syndrome and Smith-Lemli-Opitz syndrome)
Prognosis
Recent advances in medical practice, the development of surgical techniques for congenital disabilities, and improvements in health management have led to substantial improvements in survival and life expectancy among individuals with Down syndrome. A study from Birmingham, United Kingdom, conducted nearly 60 years ago, reported that only 45% of affected infants survived their first year of life and that approximately 40% were alive at age 5.[72]
In contrast, a study conducted about 50 years later found that 78% of infants with Down syndrome and congenital heart defect survived to 1 year, while survival reached 96% among those without cardiac anomalies.[73] This rise in life expectancy is expected to increase significantly due to advances in medical science. Modern healthcare systems aim to deliver timely, comprehensive, and coordinated care that enables individuals with Down syndrome to achieve optimal health, functional independence, and quality of life. While many individuals require varying degrees of support in adulthood and may not live independently, appropriate medical care, educational support, vocational training, and community resources enable most to participate meaningfully in family life, employment, and society.[74]
Complications
Infants with Down syndrome are at increased risk for several medical complications during the neonatal period and early infancy, as mentioned below.
Neonatal/Early Infancy
-
Congenital heart defect (eg, AVSD, VSD, or ASD, and PDA) or pulmonary hypertension
-
Feeding and airway issues, including hypotonia, laryngomalacia, aspiration, or GERD
-
Duodenal atresia or stenosis, as well as Hirschsprung disease
-
Transient abnormal myelopoiesis, neonatal polycythemia, or thrombocytopenia
-
Hearing loss, congenital cataracts, or nasolacrimal obstruction
-
Hypothyroidism (congenital), hypothermia risk, prolonged neonatal jaundice
Childhood/Adolescence
-
Obstructive sleep apnea (often residual after adenotonsillectomy)
-
Recurrent otitis media with effusion and/or conductive or sensorineural hearing loss
-
Refractive errors, strabismus, or amblyopia
-
Persistent feeding difficulties, chronic constipation, or celiac disease
-
Autoimmune thyroiditis
-
Leukemias (myeloid leukemia associated with Down syndrome/AMKL or Down syndrome associated with ALL)
-
Speech and language impairment, learning difficulties, attention-deficit/hyperactivity disorder, autism traits, anxiety, or depression
-
Dental caries, early periodontal disease, or bruxism
Adulthood
-
Persistent obstructive sleep apnea
-
Hypothyroidism, metabolic syndrome, dyslipidemia, or low bone density or osteoporosis
-
Progressive hearing and vision loss, keratoconus, or cataracts
-
Periodontal disease or tooth loss
-
Cervical spondylosis or stenosis with associated myelopathy
-
Early-onset Alzheimer-type dementia, depression, or anxiety
Deterrence and Patient Education
Educating families about Down syndrome is essential for reducing stress and promoting proactive, effective care. Parents should understand that Down syndrome is a genetic condition—not a disease that can be “cured”—and that with appropriate support, most children lead active lives, attend school, and participate socially. Outcomes have improved significantly compared with previous decades, with early intervention (such as physical and occupational therapies and speech-language pathology), inclusive education, and regular health monitoring being key contributors to this success.
The healthcare team should develop a clear and comprehensive plan before the newborn is discharged from the hospital. This plan should include echocardiography, newborn hearing screening with scheduled audiology follow-up, thyroid function testing at birth and again at 6 and 12 months, ophthalmology evaluation in early infancy, enrollment in early-intervention services, and information on family support groups. Clinicians should reassure families that Down syndrome occurs due to genetic factors and is not the result of anything they did or failed to do.
Practical anticipatory guidance helps prevent complications and supports effective management. Families should understand that congenital heart disease is usually treatable with appropriate cardiology care, sleep-disordered breathing is common and warrants at least one early sleep study, and routine hearing, vision, and dental evaluations are essential. Clear instructions should be provided for recognizing signs and symptoms that require prompt medical attention.
Ongoing follow-up with primary care clinicians and relevant subspecialists is crucial. Families should receive genetic counseling to discuss recurrence risk, especially in cases involving chromosomal translocations, and be connected with local and national Down syndrome support organizations. With coordinated multidisciplinary care, structured surveillance, and timely interventions, long-term outcomes are generally favorable. As families gain experience managing day-to-day care, their confidence and competence typically increase over time.
Enhancing Healthcare Team Outcomes
Individuals with Down syndrome benefit from a coordinated, interprofessional approach to care. Newborns with suspected Down syndrome should undergo karyotype analysis to confirm the diagnosis, and families should be referred to a clinical geneticist for diagnostic evaluation, parental genetic testing, and counseling.
Because Down syndrome affects multiple organ systems, ongoing care typically involves collaboration among primary care clinicians, cardiologists, ophthalmologists, otorhinolaryngologists, gastroenterologists, orthopedic surgeons, dermatologists, physical and occupational therapists, speech-language pathologists, mental health professionals, and behavioral specialists.
Parental education is central to management. Clinicians should inform families about the range of medical, developmental, and behavioral conditions associated with Down syndrome to facilitate early recognition, timely evaluation, and appropriate intervention. Management is primarily supportive and symptom-directed, aiming to optimize health, development, and functional outcomes rather than to achieve a cure.
Although life expectancy for individuals with Down syndrome has increased significantly over the past several decades, it remains lower than that of the general population. Nurses and allied health professionals play a pivotal role in care coordination, longitudinal monitoring, family education, developmental support, and early identification of emerging medical or psychosocial concerns. Their ongoing involvement, in collaboration with physicians and specialists, is essential for improving quality of life, preventing complications, and maximizing long-term outcomes for individuals with Down syndrome.
Media
(Click Image to Enlarge)

Eyes of an Infant With Down Syndrome. A cropped image of an infant’s eyes with Down syndrome, highlighting Brushfield spots visible between the inner and outer circles of the iris.
Szymon Tomczak, Public Domain, via Wikimedia Commons
(Click Image to Enlarge)
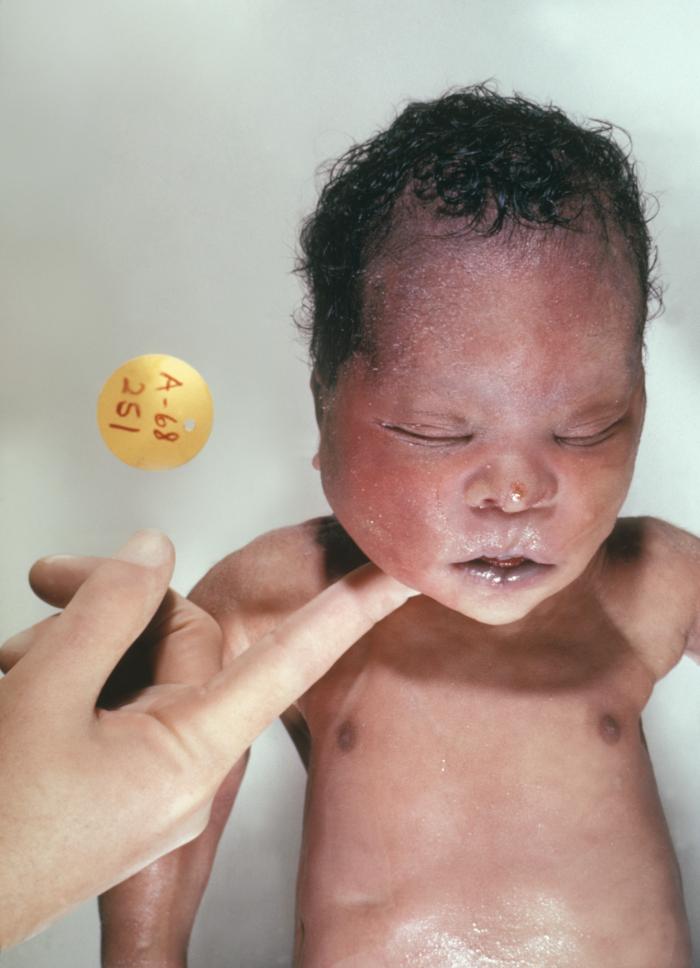
Down Syndrome. This photograph depicts a newborn with the genetic disorder Down Syndrome, due to the presence of an extra 21st chromosome.
GP Oakley, MD, Centers for Disease Control and Prevention
(Click Image to Enlarge)
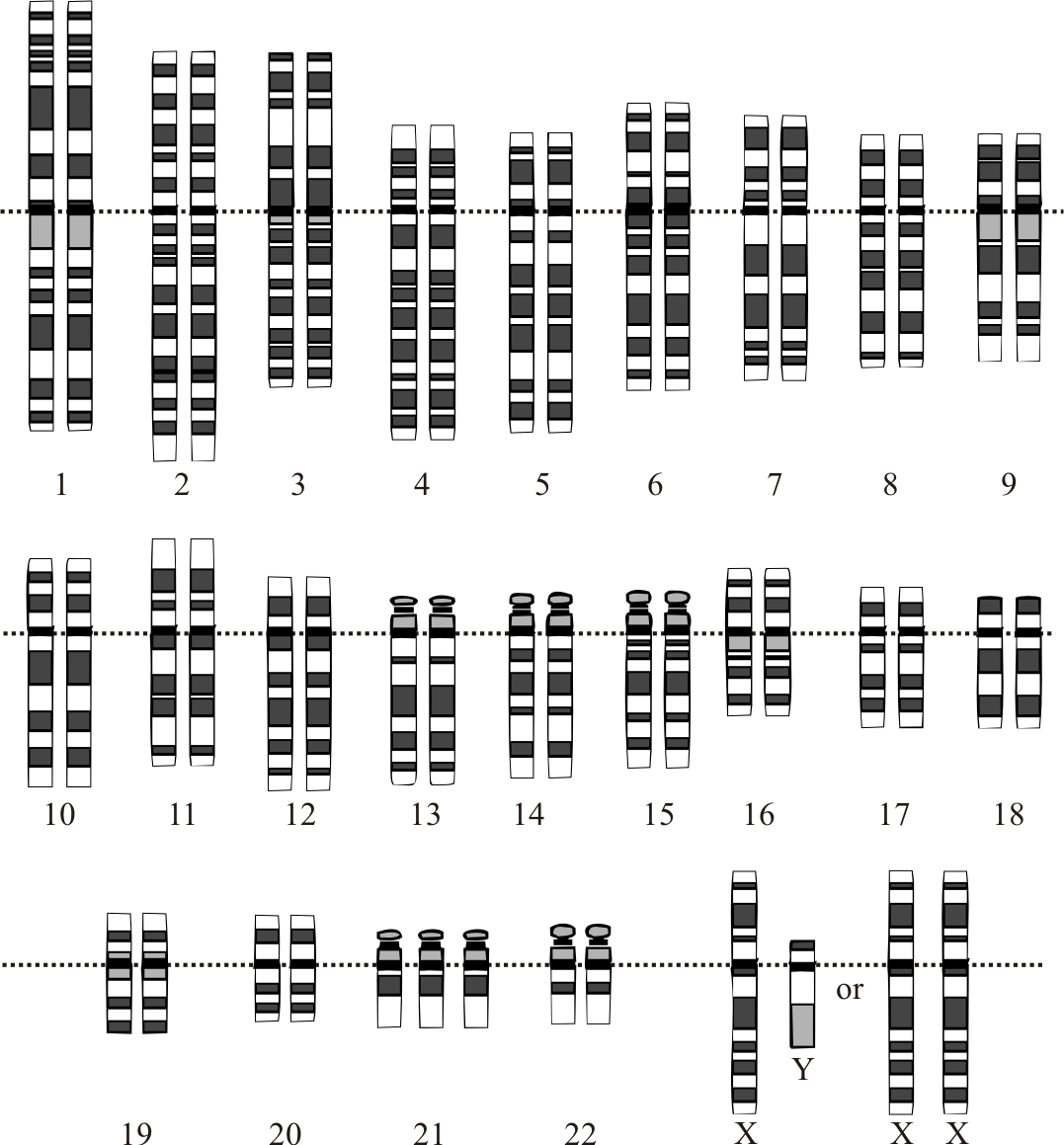
Karyotype of Down Syndrome (Trisomy 21). Karyotype displaying trisomy 21, with 3 copies of chromosome 21 clearly visible.
National Human Genome Research Institute, Human Genome Project
(Click Image to Enlarge)
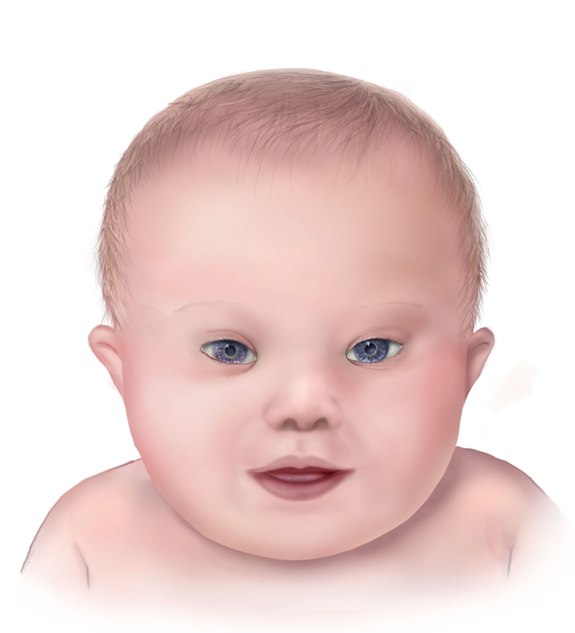
Facial Features of a Child With Down Syndrome. The illustrated image highlights the characteristic facial features of a child with Down syndrome.
Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities
(Click Image to Enlarge)
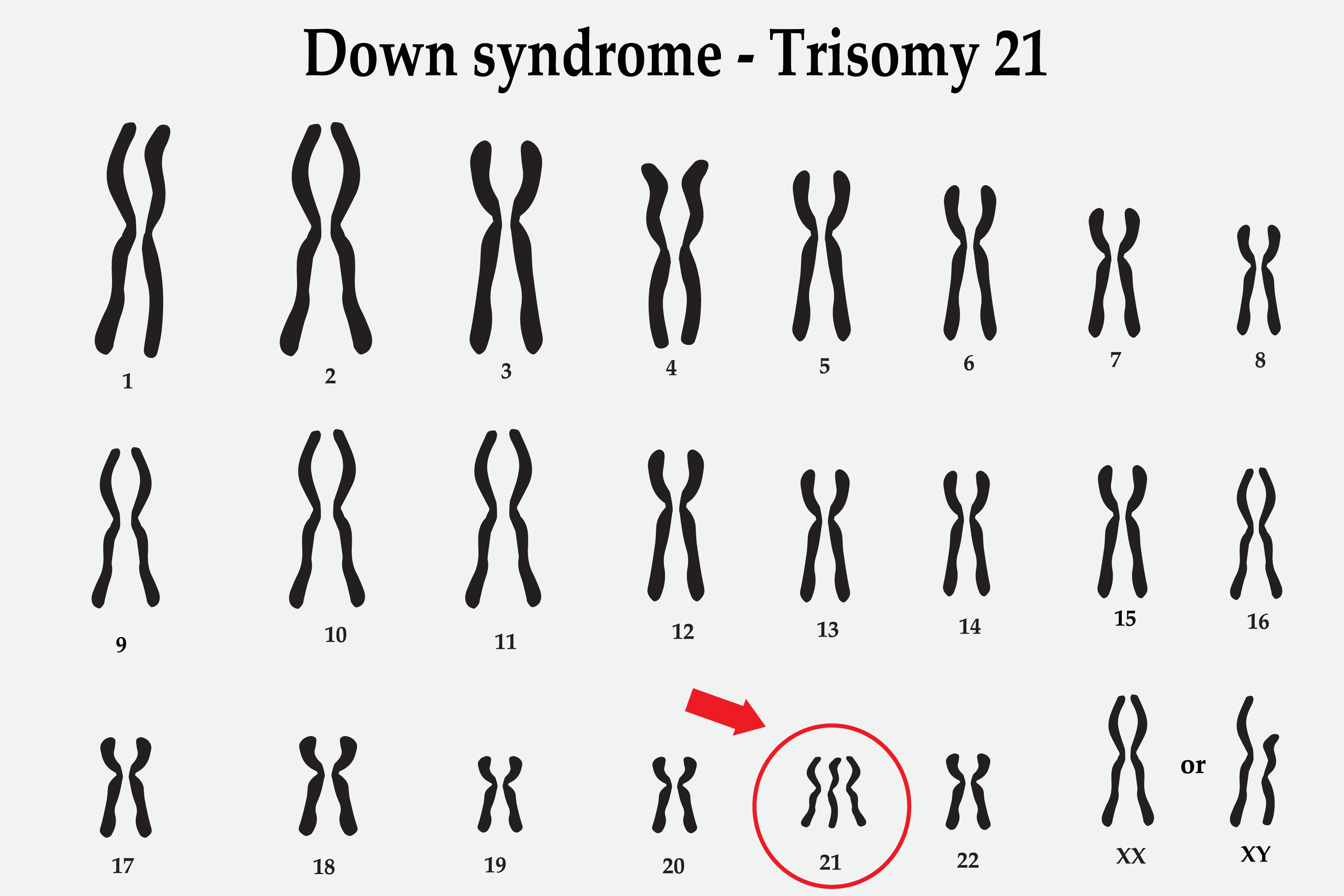
Down Syndrome (Trisomy 21) Karyotype.
Shutterstock
References
Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, Sherman SL, Reeves RH. Down syndrome. Nature reviews. Disease primers. 2020 Feb 6:6(1):9. doi: 10.1038/s41572-019-0143-7. Epub 2020 Feb 6 [PubMed PMID: 32029743]
Holmes G. Gastrointestinal disorders in Down syndrome. Gastroenterology and hepatology from bed to bench. 2014 Winter:7(1):6-8 [PubMed PMID: 25436092]
Gardiner K, Herault Y, Lott IT, Antonarakis SE, Reeves RH, Dierssen M. Down syndrome: from understanding the neurobiology to therapy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010 Nov 10:30(45):14943-5. doi: 10.1523/JNEUROSCI.3728-10.2010. Epub [PubMed PMID: 21068296]
Level 3 (low-level) evidenceHickey F, Hickey E, Summar KL. Medical update for children with Down syndrome for the pediatrician and family practitioner. Advances in pediatrics. 2012:59(1):137-57. doi: 10.1016/j.yapd.2012.04.006. Epub [PubMed PMID: 22789577]
Level 3 (low-level) evidenceBull MJ, Committee on Genetics. Health supervision for children with Down syndrome. Pediatrics. 2011 Aug:128(2):393-406. doi: 10.1542/peds.2011-1605. Epub 2011 Jul 25 [PubMed PMID: 21788214]
Zhou Y, Sheehan R, Guo L, Strydom A. Blood-based biomarkers for Alzheimer's disease in Down syndrome: A systematic review and meta-analysis. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2025 Apr:21(4):e70135. doi: 10.1002/alz.70135. Epub [PubMed PMID: 40219863]
Level 1 (high-level) evidenceHom B, Boyd NK, Vogel BN, Nishimori N, Khoshnood MM, Jafarpour S, Nagesh D, Santoro JD. Down Syndrome and Autoimmune Disease. Clinical reviews in allergy & immunology. 2024 Jun:66(3):261-273. doi: 10.1007/s12016-024-08996-2. Epub 2024 Jun 24 [PubMed PMID: 38913142]
Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nature reviews. Genetics. 2004 Oct:5(10):725-38 [PubMed PMID: 15510164]
Level 3 (low-level) evidencePritchard MA, Kola I. The "gene dosage effect" hypothesis versus the "amplified developmental instability" hypothesis in Down syndrome. Journal of neural transmission. Supplementum. 1999:57():293-303 [PubMed PMID: 10666684]
Level 3 (low-level) evidenceHolland AJ, Hon J, Huppert FA, Stevens F. Incidence and course of dementia in people with Down's syndrome: findings from a population-based study. Journal of intellectual disability research : JIDR. 2000 Apr:44 ( Pt 2)():138-46 [PubMed PMID: 10898377]
Asim A, Kumar A, Muthuswamy S, Jain S, Agarwal S. "Down syndrome: an insight of the disease". Journal of biomedical science. 2015 Jun 11:22(1):41. doi: 10.1186/s12929-015-0138-y. Epub 2015 Jun 11 [PubMed PMID: 26062604]
Bittles AH, Glasson EJ. Clinical, social, and ethical implications of changing life expectancy in Down syndrome. Developmental medicine and child neurology. 2004 Apr:46(4):282-6 [PubMed PMID: 15077706]
Roper RJ, Reeves RH. Understanding the basis for Down syndrome phenotypes. PLoS genetics. 2006 Mar:2(3):e50 [PubMed PMID: 16596169]
Level 3 (low-level) evidenceChaiken SR, Mandelbaum AD, Garg B, Doshi U, Packer CH, Caughey AB. Association Between Rates of Down Syndrome Diagnosis in States With vs Without 20-Week Abortion Bans From 2011 to 2018. JAMA network open. 2023 Mar 1:6(3):e233684. doi: 10.1001/jamanetworkopen.2023.3684. Epub 2023 Mar 1 [PubMed PMID: 36943268]
Halder P, Pal U, Ganguly A, Ghosh P, Ray A, Sarkar S, Ghosh S. Genetic aetiology of Down syndrome birth: novel variants of maternal DNMT3B and RFC1 genes increase risk of meiosis II nondisjunction in the oocyte. Molecular genetics and genomics : MGG. 2023 Jan:298(1):293-313. doi: 10.1007/s00438-022-01981-4. Epub 2022 Nov 30 [PubMed PMID: 36447056]
Blanco-Montaño A, Ramos-Arenas M, Yerena-Echevarría BA, Miranda-Santizo LD, Ríos-Celis AL, Dorantes-Gómez AT, Morato-Rangel AJ, Meza-Hernández JA, Acosta-Saldívar ED, Aguilar-Castillo CD, Cárdenas-Conejo A. [Risk factors in the origin of Down syndrome]. Revista medica del Instituto Mexicano del Seguro Social. 2023 Sep 4:61(5):638-644. doi: 10.5281/zenodo.8316459. Epub 2023 Sep 4 [PubMed PMID: 37769135]
Choi JK. Hematopoietic disorders in Down syndrome. International journal of clinical and experimental pathology. 2008 Jan 1:1(5):387-95 [PubMed PMID: 18787621]
Benhaourech S, Drighil A, Hammiri AE. Congenital heart disease and Down syndrome: various aspects of a confirmed association. Cardiovascular journal of Africa. 2016 Sep/Oct:27(5):287-290. doi: 10.5830/CVJA-2016-019. Epub [PubMed PMID: 27805241]
Wiseman FK, Alford KA, Tybulewicz VL, Fisher EM. Down syndrome--recent progress and future prospects. Human molecular genetics. 2009 Apr 15:18(R1):R75-83. doi: 10.1093/hmg/ddp010. Epub [PubMed PMID: 19297404]
Level 3 (low-level) evidenceDimopoulos K, Constantine A, Clift P, Condliffe R, Moledina S, Jansen K, Inuzuka R, Veldtman GR, Cua CL, Tay ELW, Opotowsky AR, Giannakoulas G, Alonso-Gonzalez R, Cordina R, Capone G, Namuyonga J, Scott CH, D'Alto M, Gamero FJ, Chicoine B, Gu H, Limsuwan A, Majekodunmi T, Budts W, Coghlan G, Broberg CS, for Down Syndrome International (DSi). Cardiovascular Complications of Down Syndrome: Scoping Review and Expert Consensus. Circulation. 2023 Jan 31:147(5):425-441. doi: 10.1161/CIRCULATIONAHA.122.059706. Epub 2023 Jan 30 [PubMed PMID: 36716257]
Level 2 (mid-level) evidenceAmiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, Brooks AS, Antinolo G, de Pontual L, Clement-Ziza M, Munnich A, Kashuk C, West K, Wong KK, Lyonnet S, Chakravarti A, Tam PK, Ceccherini I, Hofstra RM, Fernandez R, Hirschsprung Disease Consortium. Hirschsprung disease, associated syndromes and genetics: a review. Journal of medical genetics. 2008 Jan:45(1):1-14 [PubMed PMID: 17965226]
Elgendy MM, Cortez J, Saker F, Mohamed MA, Aly H. Prevalence and Outcomes of Gastrointestinal Anomalies in Down Syndrome. American journal of perinatology. 2024 Nov:41(15):2047-2052. doi: 10.1055/s-0044-1786874. Epub 2024 May 14 [PubMed PMID: 38744322]
Wallace RA. Clinical audit of gastrointestinal conditions occurring among adults with Down syndrome attending a specialist clinic. Journal of intellectual & developmental disability. 2007 Mar:32(1):45-50 [PubMed PMID: 17365367]
Henry E, Walker D, Wiedmeier SE, Christensen RD. Hematological abnormalities during the first week of life among neonates with Down syndrome: data from a multihospital healthcare system. American journal of medical genetics. Part A. 2007 Jan 1:143A(1):42-50 [PubMed PMID: 17163522]
Hord JD, Gay JC, Whitlock JA. Thrombocytopenia in neonates with trisomy 21. Archives of pediatrics & adolescent medicine. 1995 Jul:149(7):824-5 [PubMed PMID: 7795778]
Level 2 (mid-level) evidenceMiller M, Cosgriff JM. Hematological abnormalities in newborn infants with Down syndrome. American journal of medical genetics. 1983 Oct:16(2):173-7 [PubMed PMID: 6228141]
Zipursky A, Brown E, Christensen H, Sutherland R, Doyle J. Leukemia and/or myeloproliferative syndrome in neonates with Down syndrome. Seminars in perinatology. 1997 Feb:21(1):97-101 [PubMed PMID: 9190039]
Zipursky A, Brown EJ, Christensen H, Doyle J. Transient myeloproliferative disorder (transient leukemia) and hematologic manifestations of Down syndrome. Clinics in laboratory medicine. 1999 Mar:19(1):157-67, vii [PubMed PMID: 10403079]
Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet (London, England). 2000 Jan 15:355(9199):165-9 [PubMed PMID: 10675114]
Verma A, Lupo PJ, Shah NN, Hitzler J, Rabin KR. Management of Down Syndrome-Associated Leukemias: A Review. JAMA oncology. 2023 Sep 1:9(9):1283-1290. doi: 10.1001/jamaoncol.2023.2163. Epub [PubMed PMID: 37440251]
Kearney L, Gonzalez De Castro D, Yeung J, Procter J, Horsley SW, Eguchi-Ishimae M, Bateman CM, Anderson K, Chaplin T, Young BD, Harrison CJ, Kempski H, So CW, Ford AM, Greaves M. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009 Jan 15:113(3):646-8. doi: 10.1182/blood-2008-08-170928. Epub 2008 Oct 16 [PubMed PMID: 18927438]
Level 3 (low-level) evidenceBarwe SP, Kolb EA, Gopalakrishnapillai A. Down syndrome and leukemia: An insight into the disease biology and current treatment options. Blood reviews. 2024 Mar:64():101154. doi: 10.1016/j.blre.2023.101154. Epub 2023 Nov 25 [PubMed PMID: 38016838]
Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nature genetics. 2002 Sep:32(1):148-52 [PubMed PMID: 12172547]
Pearlson GD, Breiter SN, Aylward EH, Warren AC, Grygorcewicz M, Frangou S, Barta PE, Pulsifer MB. MRI brain changes in subjects with Down syndrome with and without dementia. Developmental medicine and child neurology. 1998 May:40(5):326-34 [PubMed PMID: 9630260]
Lott IT. Neurological phenotypes for Down syndrome across the life span. Progress in brain research. 2012:197():101-21. doi: 10.1016/B978-0-444-54299-1.00006-6. Epub [PubMed PMID: 22541290]
Agiovlasitis S, McCubbin JA, Yun J, Pavol MJ, Widrick JJ. Economy and preferred speed of walking in adults with and without Down syndrome. Adapted physical activity quarterly : APAQ. 2009 Apr:26(2):118-30 [PubMed PMID: 19478345]
Rubenstein E, Tewolde S, Michals A, Weuve J, Fortea J, Fox MP, Pescador Jimenez M, Scott A, Tripodis Y, Skotko BG. Alzheimer Dementia Among Individuals With Down Syndrome. JAMA network open. 2024 Sep 3:7(9):e2435018. doi: 10.1001/jamanetworkopen.2024.35018. Epub 2024 Sep 3 [PubMed PMID: 39312235]
Merrick J, Ezra E, Josef B, Hendel D, Steinberg DM, Wientroub S. Musculoskeletal problems in Down Syndrome European Paediatric Orthopaedic Society Survey: the Israeli sample. Journal of pediatric orthopedics. Part B. 2000 Jun:9(3):185-92 [PubMed PMID: 10904905]
Level 3 (low-level) evidenceArya R, Kabra M, Gulati S. Epilepsy in children with Down syndrome. Epileptic disorders : international epilepsy journal with videotape. 2011 Mar:13(1):1-7. doi: 10.1684/epd.2011.0415. Epub [PubMed PMID: 21398208]
Pueschel SM, Louis S, McKnight P. Seizure disorders in Down syndrome. Archives of neurology. 1991 Mar:48(3):318-20 [PubMed PMID: 1825777]
Ferlazzo E, Adjien CK, Guerrini R, Calarese T, Crespel A, Elia M, Striano P, Gelisse P, Bramanti P, di Bella P, Genton P. Lennox-Gastaut syndrome with late-onset and prominent reflex seizures in trisomy 21 patients. Epilepsia. 2009 Jun:50(6):1587-95. doi: 10.1111/j.1528-1167.2008.01944.x. Epub 2009 Jan 31 [PubMed PMID: 19187280]
Level 2 (mid-level) evidenceMenéndez M. Down syndrome, Alzheimer's disease and seizures. Brain & development. 2005 Jun:27(4):246-52 [PubMed PMID: 15862185]
De Simone R, Puig XS, Gélisse P, Crespel A, Genton P. Senile myoclonic epilepsy: delineation of a common condition associated with Alzheimer's disease in Down syndrome. Seizure. 2010 Sep:19(7):383-9. doi: 10.1016/j.seizure.2010.04.008. Epub 2010 Jul 3 [PubMed PMID: 20598585]
Level 3 (low-level) evidenceLott IT, Doran E, Nguyen VQ, Tournay A, Movsesyan N, Gillen DL. Down syndrome and dementia: seizures and cognitive decline. Journal of Alzheimer's disease : JAD. 2012:29(1):177-85. doi: 10.3233/JAD-2012-111613. Epub [PubMed PMID: 22214782]
Level 2 (mid-level) evidenceJanicki MP, Dalton AJ. Prevalence of dementia and impact on intellectual disability services. Mental retardation. 2000 Jun:38(3):276-88 [PubMed PMID: 10900935]
Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Annals of neurology. 2004 Nov:56(5):675-88 [PubMed PMID: 15468085]
Level 3 (low-level) evidenceVoronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, Arancio O, Davisson MT, Antonarakis SE, Gardiner K, De Camilli P, Di Paolo G. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down's syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2008 Jul 8:105(27):9415-20. doi: 10.1073/pnas.0803756105. Epub 2008 Jun 30 [PubMed PMID: 18591654]
Level 3 (low-level) evidenceHawli Y, Nasrallah M, El-Hajj Fuleihan G. Endocrine and musculoskeletal abnormalities in patients with Down syndrome. Nature reviews. Endocrinology. 2009 Jun:5(6):327-34. doi: 10.1038/nrendo.2009.80. Epub [PubMed PMID: 19421241]
Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, O'Heir CE, Mitchell ML, Hermos RJ, Waisbren SE, Faix JD, Klein RZ. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. The New England journal of medicine. 1999 Aug 19:341(8):549-55 [PubMed PMID: 10451459]
Level 2 (mid-level) evidenceTüysüz B, Beker DB. Thyroid dysfunction in children with Down's syndrome. Acta paediatrica (Oslo, Norway : 1992). 2001 Dec:90(12):1389-93 [PubMed PMID: 11853334]
Lavard L, Ranløv I, Perrild H, Andersen O, Jacobsen BB. Incidence of juvenile thyrotoxicosis in Denmark, 1982-1988. A nationwide study. European journal of endocrinology. 1994 Jun:130(6):565-8 [PubMed PMID: 8205255]
Thomas JR, Roper RJ. Current Analysis of Skeletal Phenotypes in Down Syndrome. Current osteoporosis reports. 2021 Jun:19(3):338-346. doi: 10.1007/s11914-021-00674-y. Epub 2021 Apr 8 [PubMed PMID: 33830429]
Morris AF, Vaughan SE, Vaccaro P. Measurements of neuromuscular tone and strength in Down's syndrome children. Journal of mental deficiency research. 1982 Mar:26(Pt 1):41-6 [PubMed PMID: 6210779]
Cabana MD, Capone G, Fritz A, Berkovitz G. Nutritional rickets in a child with Down syndrome. Clinical pediatrics. 1997 Apr:36(4):235-7 [PubMed PMID: 9114996]
Level 3 (low-level) evidenceMerrick J, Koslowe K. Refractive errors and visual anomalies in Down syndrome. Down's syndrome, research and practice : the journal of the Sarah Duffen Centre. 2001 Jul:6(3):131-3 [PubMed PMID: 11501216]
Shott SR. Down syndrome: common otolaryngologic manifestations. American journal of medical genetics. Part C, Seminars in medical genetics. 2006 Aug 15:142C(3):131-40 [PubMed PMID: 16838306]
Agathokleous M, Chaveeva P, Poon LC, Kosinski P, Nicolaides KH. Meta-analysis of second-trimester markers for trisomy 21. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology. 2013 Mar:41(3):247-61. doi: 10.1002/uog.12364. Epub 2013 Jan 24 [PubMed PMID: 23208748]
Level 1 (high-level) evidenceRenna MD, Pisani P, Conversano F, Perrone E, Casciaro E, Renzo GC, Paola MD, Perrone A, Casciaro S. Sonographic markers for early diagnosis of fetal malformations. World journal of radiology. 2013 Oct 28:5(10):356-71. doi: 10.4329/wjr.v5.i10.356. Epub [PubMed PMID: 24179631]
Hui L, Pynaker C, Bonacquisto L, Lindquist A, Poulton A, Kluckow E, Hutchinson B, Norris F, Pertile MD, Gugasyan L, Kulkarni A, Harraway J, Howden A, McCoy R, da Silva Costa F, Menezes M, Palma-Dias R, Nisbet D, Martin N, Bethune M, Poulakis Z, Halliday J. Reexamining the optimal nuchal translucency cutoff for diagnostic testing in the cell-free DNA and microarray era: results from the Victorian Perinatal Record Linkage study. American journal of obstetrics and gynecology. 2021 Nov:225(5):527.e1-527.e12. doi: 10.1016/j.ajog.2021.03.050. Epub 2021 May 3 [PubMed PMID: 33957116]
Kuo WL, Tenjin H, Segraves R, Pinkel D, Golbus MS, Gray J. Detection of aneuploidy involving chromosomes 13, 18, or 21, by fluorescence in situ hybridization (FISH) to interphase and metaphase amniocytes. American journal of human genetics. 1991 Jul:49(1):112-9 [PubMed PMID: 2063863]
Jain S, Agarwal S, Panigrahi I, Tamhankar P, Phadke S. Diagnosis of Down syndrome and detection of origin of nondisjunction by short tandem repeat analysis. Genetic testing and molecular biomarkers. 2010 Aug:14(4):489-91. doi: 10.1089/gtmb.2009.0191. Epub [PubMed PMID: 20722466]
Level 2 (mid-level) evidenceJain S, Panigrahi I, Gupta R, Phadke SR, Agarwal S. Multiplex quantitative fluorescent polymerase chain reaction for detection of aneuploidies. Genetic testing and molecular biomarkers. 2012 Jun:16(6):624-7. doi: 10.1089/gtmb.2011.0243. Epub 2012 Feb 7 [PubMed PMID: 22313045]
Deutsch S, Choudhury U, Merla G, Howald C, Sylvan A, Antonarakis SE. Detection of aneuploidies by paralogous sequence quantification. Journal of medical genetics. 2004 Dec:41(12):908-15 [PubMed PMID: 15591276]
Walknowska J, Conte FA, Grumbach MM. Practical and theoretical implications of fetal-maternal lymphocyte transfer. Lancet (London, England). 1969 Jun 7:1(7606):1119-22 [PubMed PMID: 4181601]
Daniels G, Finning K, Martin P, Massey E. Noninvasive prenatal diagnosis of fetal blood group phenotypes: current practice and future prospects. Prenatal diagnosis. 2009 Feb:29(2):101-7. doi: 10.1002/pd.2172. Epub [PubMed PMID: 19085963]
Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, Wainscoat JS. Presence of fetal DNA in maternal plasma and serum. Lancet (London, England). 1997 Aug 16:350(9076):485-7 [PubMed PMID: 9274585]
Level 2 (mid-level) evidenceWright CF, Burton H. The use of cell-free fetal nucleic acids in maternal blood for non-invasive prenatal diagnosis. Human reproduction update. 2009 Jan-Feb:15(1):139-51. doi: 10.1093/humupd/dmn047. Epub 2008 Oct 22 [PubMed PMID: 18945714]
Dungan JS, Klugman S, Darilek S, Malinowski J, Akkari YMN, Monaghan KG, Erwin A, Best RG, ACMG Board of Directors. Electronic address: documents@acmg.net. Noninvasive prenatal screening (NIPS) for fetal chromosome abnormalities in a general-risk population: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genetics in medicine : official journal of the American College of Medical Genetics. 2023 Feb:25(2):100336. doi: 10.1016/j.gim.2022.11.004. Epub 2022 Dec 16 [PubMed PMID: 36524989]
Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clinical chemistry. 2009 Apr:55(4):641-58. doi: 10.1373/clinchem.2008.112789. Epub 2009 Feb 26 [PubMed PMID: 19246620]
Level 3 (low-level) evidenceBull MJ, Trotter T, Santoro SL, Christensen C, Grout RW, COUNCIL ON GENETICS, Burke LW, Berry SA, Geleske TA, Holm I, Hopkin RJ, Introne WJ, Lyons MJ, Monteil DC, Scheuerle A, Stoler JM, Vergano SA, Chen E, Hamid R, Downs SM, Grout RW, Cunniff C, Parisi MA, Ralston SJ, Scott JA, Shapira SK, Spire P. Health Supervision for Children and Adolescents With Down Syndrome. Pediatrics. 2022 May 1:149(5):. pii: e2022057010. doi: 10.1542/peds.2022-057010. Epub [PubMed PMID: 35490285]
Bunt CW, Bunt SK. Care of Infants and Children With Down Syndrome: Role of the Family Physician. American family physician. 2025 Mar:111(3):236-244 [PubMed PMID: 40106290]
RECORD RG, SMITH A. Incidence, mortality, and dex distribution of mongoloid defectives. British journal of preventive & social medicine. 1955 Jan:9(1):10-5 [PubMed PMID: 14351707]
Bell R, Rankin J, Donaldson LJ, Northern Congenital Abnormality Survey Steering Group. Down's syndrome: occurrence and outcome in the north of England, 1985-99. Paediatric and perinatal epidemiology. 2003 Jan:17(1):33-9 [PubMed PMID: 12562470]
Skotko BG, Davidson EJ, Weintraub GS. Contributions of a specialty clinic for children and adolescents with Down syndrome. American journal of medical genetics. Part A. 2013 Mar:161A(3):430-7. doi: 10.1002/ajmg.a.35795. Epub 2013 Feb 7 [PubMed PMID: 23401090]
Level 2 (mid-level) evidence