Introduction
Interindividual variability in drug response remains a major challenge in clinical therapy, with outcomes ranging from treatment failure to serious adverse drug reactions. A significant proportion of this variability arises from differences in drug metabolism, much of which is governed by the cytochrome P450 (CYP) enzyme system. Predominantly expressed in hepatocytes, CYP enzymes catalyze the biotransformation and clearance of a wide array of xenobiotics and potentially toxic compounds. Although these enzymes participate in essential physiological processes, including steroid and hormone synthesis, metabolism of fat-soluble vitamins, and regulation of fatty acids, their most critical impact lies in pharmacokinetics and drug metabolism. A relatively small subset of CYP isoforms accounts for the majority of drug-processing activity, with CYP family enzymes collectively involved in an estimated 80% to 90% of enzymatic drug metabolism.[1] Because drugs can either influence CYP activity or be influenced by it, unintended interactions may arise, altering therapeutic efficacy or increasing toxicity. Understanding how drugs modulate CYP enzymes and how genetic, environmental, and pharmacological factors shape CYP function is essential for optimizing therapy and ensuring safe, effective clinical outcomes.[2]
Fundamentals
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Fundamentals
CYP enzymes are mixed-function oxidases that require both NADPH (nicotinamide adenine dinucleotide phosphate) and molecular oxygen to carry out their reactions. These enzymes participate in numerous biosynthetic pathways, including key steps in the conversion of lanosterol to cholesterol and in the synthesis of steroid hormones. CYP enzymes are also essential for the detoxification of xenobiotics and the metabolism of many therapeutic drugs. CYP represents a large superfamily of membrane-bound hemoprotein isozymes. Although these enzymes are present throughout the body, they are most abundant in the liver, with significant activity also found in the intestines and kidneys. Although humans possess 57 functional CYP genes, the majority of drug metabolism is mediated by a small subset of approximately 6 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4), accounting for an estimated 80% to 90% of reactions.[3] Because different drugs are processed by different CYP isoforms, identifying which enzymes are involved is critical in both drug development and clinical practice.
Understanding CYP induction and inhibition is essential in predicting drug interactions. Most medications undergo metabolic inactivation before being excreted. In general, CYP enzymes convert lipophilic substrates into more hydrophilic metabolites to facilitate their elimination.[4] If a drug induces a CYP enzyme, it increases the metabolism of other drugs that use the same pathway, potentially lowering their plasma concentrations below therapeutic levels and leading to treatment failure. Conversely, CYP inhibition slows metabolism, increasing the risk of accumulation, toxicity, and adverse effects of other drugs using the affected pathway. These principles differ in the case of prodrugs, which require metabolic activation. Genetic differences further influence CYP activity. Individuals may be classified as poor, intermediate, extensive, or ultrarapid metabolizers depending on inherited variations in specific CYP genes. These phenotypes can significantly affect drug response and safety, underscoring the importance of personalized dosing and careful therapeutic monitoring.[5][6]
Issues of Concern
CYP enzymes play a central role in the biotransformation of xenobiotics and are widely expressed across multiple organ systems, making them subject to extensive biochemical interactions and highly relevant to pharmacological research. The influence of CYP enzymes on drug disposition, safety, and efficacy has placed them at the forefront of drug development, where characterization of CYP-mediated induction, inhibition, and substrate specificity is essential. Certain medications act as potent modulators of CYP activity, necessitating careful clinical monitoring, particularly when multiple agents dependent on the same isoforms are co-administered. Notably, CYP3A4 and CYP2D6 together account for a substantial proportion of clinically relevant drug metabolism, underscoring their importance in predicting and mitigating drug-drug interactions.[3]
Although the CYP system possesses considerable metabolic capacity—due to the ability of individual isoenzymes to accommodate and process multiple substrates at distinct active sites—its function may be compromised in the presence of hepatic comorbidities such as cirrhosis or viral hepatitis. Under these conditions, the risk of adverse drug reactions increases. Drug toxicity resulting from CYP inhibition typically manifests as symptoms consistent with overdose, and management often involves withholding the offending agent until plasma concentrations normalize, with antidotal therapy reserved for severe cases. Conversely, treatment failure attributable to accelerated drug clearance reflects enzyme induction, necessitating dose modification or selection of an alternative therapeutic agent.[3]
Prodrugs—pharmacologically inactive compounds that are metabolized into pharmacologically active drugs after intake—introduce an additional layer of complexity, as they require metabolic activation to yield pharmacologically active metabolites. Codeine, which relies on CYP2D6-mediated conversion to morphine, exemplifies this phenomenon. Individuals who are genetically poor metabolizers exhibit minimal therapeutic response due to insufficient bioactivation, whereas ultrarapid metabolizers generate elevated morphine concentrations that can precipitate life-threatening respiratory depression. These genotype-dependent differences highlight the clinical importance of integrating pharmacogenetic data into therapeutic decision-making.[6]
The expanding use of herbal supplements further complicates the clinical landscape, given the limited regulatory oversight and incomplete characterization of their pharmacologic effects. Several natural products modulate CYP activity, potentially significantly influencing drug exposure. St. John's Wort is a well-documented inducer of CYP3A4 and can markedly decrease plasma concentrations of co-administered medications. In contrast, grapefruit juice acts as a CYP3A4 inhibitor, elevating drug levels and increasing toxicity risk. Nicotine induces CYP1A2, and both nicotine and caffeine serve as substrates of this isoform, offering a mechanistic basis for the heightened caffeine tolerance observed in individuals with chronic cigarette use. Such interactions emphasize the need for comprehensive medication and substance use histories that include over-the-counter and herbal products.[7][8]
Cellular Level
Within cells, CYP enzymes are predominantly localized to the endoplasmic reticulum and the inner mitochondrial membrane, where they participate in distinct metabolic pathways. Endoplasmic reticulum–associated CYP isoforms primarily catalyze the biotransformation of xenobiotics, including pharmaceutical agents and environmental chemicals. In contrast, mitochondrial CYP enzymes are more directly involved in endogenous metabolic processes such as steroid hormone biosynthesis, cholesterol metabolism, and the regulation of fatty acid oxidation. CYP activity is tightly regulated at the transcriptional level, as demonstrated by the correlation between enzyme abundance and corresponding messenger RNA expression profiles.[9]
The primary purpose of drug metabolism is to facilitate the safe elimination of exogenous compounds by converting lipophilic substances into more hydrophilic metabolites. Once rendered water-soluble, drugs and their metabolites are efficiently transported to the kidneys for filtration and excretion. Classically, drug metabolism is divided into 3 phases, with phase I reactions—mediated largely by the CYP system—representing the initial biochemical modifications. These reactions include oxidation, reduction, and hydrolysis, with oxidation serving as the predominant mechanism through which substrates undergo structural transformation.[10][11]
Molecular Level
CYP enzymes are heme-containing monooxygenases, typically composed of approximately 400 to 500 amino acids, and are characterized by a single heme prosthetic group embedded within the active site.[12] To date, more than 100 distinct CYP structures have been resolved and deposited in the Protein Data Bank. Despite substantial sequence variability among isoforms, CYP enzymes share a conserved tertiary architecture consisting of a predominantly α-helical fold—commonly described as containing around 12 major helices—supported by several β-sheet regions. This structural framework permits considerable conformational flexibility, enabling the active site to accommodate substrates of diverse sizes and shapes, thereby accounting for the broad substrate specificity characteristic of CYP isoenzymes.[12]
CYP genetic variation contributes significantly to interindividual differences in drug metabolism. Alleles are broadly classified into normal-function, decreased- or no-function, and increased-function variants, rather than strictly wild type versus variant. Individuals carrying 2 normal-function alleles are typically classified as extensive (normal) metabolizers, whereas those with 2 decreased- or no-function alleles are considered poor metabolizers. Heterozygous individuals with 1 normal and 1 reduced-function allele generally fall into the intermediate metabolizer category. Ultrarapid metabolizers arise from gene duplication or multiplication events, most notably in CYP2D6, resulting in more than 2 active gene copies and subsequently increased enzymatic activity. These pharmacogenomic differences explain why patients may experience distinct therapeutic responses or toxicity risks at standard dosages. More than 350 functional polymorphisms have been identified across the human CYP superfamily, reflecting the extensive genetic diversity that influences drug disposition.[6][8]
Beyond inherited DNA variation, CYP expression is regulated by epigenetic mechanisms and influenced by multiple physiological and environmental factors. Age, sex, hormonal status, infection, inflammation, and exposure to xenobiotics can modulate the expression or activity of specific CYP isoforms. Notably, sex-related differences in CYP expression have been reported; for example, certain CYP enzymes exhibit higher expression in females, providing a potential mechanistic basis for sex-dependent differences in drug metabolism. Although substantial progress has been made in characterizing CYP structure, function, and genetic variability, ongoing molecular research—enhanced by insights from the Human Genome Project—continues to refine our understanding of how CYP polymorphisms contribute to interindividual variability in drug response.[3] These developments have positioned CYP pharmacogenomics as a central component of precision medicine.
Function
CYP enzymes are essential for the metabolism and elimination of drugs. Among the CYP family, CYP2D6 and CYP3A4/5 are particularly notable, participating in the metabolism of approximately 20% and 30% of all clinically used drugs, respectively (see Image. Fractional Contribution of Specific CYP Isoforms to the Metabolism of 248 Drugs).[12] CYP enzymes predominantly mediate phase I biotransformation reactions of xenobiotics, including oxidation, reduction, and hydrolysis. Through these reactions, lipid-soluble compounds are converted into more water-soluble metabolites, facilitating renal or biliary excretion. However, the oxidative processes catalyzed by CYPs can generate reactive intermediates, which may contribute to cellular toxicity, mutagenesis, or carcinogenesis. Consequently, CYP-mediated metabolism can influence both the pharmacokinetics and pharmacodynamics of therapeutic agents.[13]
Beyond xenobiotic metabolism, CYP enzymes play critical roles in steroidogenesis and the biosynthesis of bile acids, eicosanoids, and fat-soluble vitamins. In endogenous metabolism, multiple CYP isoforms participate in steroidogenesis, converting cholesterol precursors into mineralocorticoids, glucocorticoids, and sex steroids, including androgens and estrogens (See Image. Steroid Biosynthesis Pathway). CYP17A1 catalyzes a key step in the synthesis of mineralocorticoids, whereas CYP21A2 is essential for the production of both mineralocorticoids and glucocorticoids. These enzymatic pathways underscore the dual significance of CYP enzymes in both xenobiotic metabolism and fundamental physiological processes.[13][14]
Mechanism
CYP enzymes catalyze a wide array of reactions, with variations determined by the specific isoform and substrate involved. As monooxygenases, their primary function is to insert a single oxygen atom into the substrate, typically resulting in hydroxylation. The catalytic mechanism of CYP enzymes follows a series of coordinated steps:
- Active site composition: The CYP active site contains a heme-iron center, with the iron coordinated to the protein via a cysteine thiolate ligand.
- Substrate binding: The substrate binds to the heme iron, inducing a conformational change that optimizes enzyme–substrate interactions.
- Electron transfer: Electrons are transferred from NAD(P)H to the heme iron via associated reductase proteins, reducing the ferric iron (Fe³+) to the ferrous state (Fe²+).
- Oxygen Activation: Molecular oxygen binds to the reduced iron, forming an iron-oxygen complex. A second electron is then delivered, generating a reactive ferric-peroxide intermediate.
- Formation of compound I: Protonation of the peroxide intermediate leads to water release and the formation of the highly reactive oxidizing species, P450 compound I (FeO³+).
- Substrate hydroxylation: Compound I abstracts a hydrogen atom from the substrate and inserts an oxygen atom, producing a hydroxylated metabolite, which is typically more hydrophilic and readily excretable.
The overall reaction can be summarized as:
O2 + NAD(P)H + H+ + RH → NAD(P)+ + H2O + ROH
Compounds that already possess polar functional groups may bypass phase I metabolism and proceed directly to phase II conjugation reactions, such as glucuronidation, sulfation, or glutathione conjugation, thereby enhancing solubility and facilitating excretion.[15][10][6]
Pathophysiology
Inflammatory states significantly influence CYP enzyme expression and activity. Hepatic diseases such as cirrhosis, characterized by progressive fibrosis and hepatocellular loss, markedly reduce metabolic capacity due to impaired hepatocyte function. The clinical manifestations of cirrhosis are closely linked to these metabolic disturbances. Altered steroid hormone metabolism, partly mediated by disrupted CYP-dependent steroidogenic pathways, contributes to the development of gynecomastia, spider angiomas, and other endocrine abnormalities commonly observed in patients with cirrhosis.[16] In addition to reduced CYP activity, cirrhosis is associated with diminished hepatic synthesis of plasma proteins, including albumin, which further impacts drug distribution and free drug concentrations.[17]
Because CYP enzymes are also expressed in the intestinal epithelium, disorders affecting the small or large bowel can alter presystemic metabolism and oral drug bioavailability. Although intestinal CYP enzymes contribute only modestly to overall xenobiotic metabolism compared to hepatic CYPs, their role in first-pass metabolism and absorption is clinically significant. Disease-related changes or concurrent exposures may therefore reduce or enhance systemic drug levels. Grapefruit juice is a well-known example of such interactions: its flavonoid component, naringin, inhibits intestinal CYP3A4, decreasing enterocyte-mediated metabolism and increasing systemic concentrations of CYP3A4 substrates even at relatively small ingested amounts.[6]
Infectious and inflammatory conditions further modulate CYP activity through cytokine-mediated downregulation. Interleukins, interferons, tumor necrosis factor, and other inflammatory mediators suppress the transcription and activity of multiple CYP isoforms.[3] This immune-driven reduction in metabolic capacity underscores the broader role of inflammation in altering pharmacokinetics during both localized and systemic infections.
Clinical Significance
A comprehensive medication review is essential for all patients undergoing drug therapy to identify potential interactions that may alter CYP enzyme activity. Depending on a compound's pharmacological profile, CYP isoforms may be either induced or inhibited, leading to clinically significant alterations in drug metabolism and clearance. Such interactions can result in drug accumulation and toxicity or, conversely, accelerated clearance and therapeutic failure. Clinicians must also consider the influence of naturally occurring compounds—such as grapefruit juice, nicotine-containing products, and St. John's wort—which are well-established modulators of CYP function. Additional caution is warranted in patients with hepatic injury or reduced baseline CYP activity, as these conditions amplify the risk of adverse drug reactions.
Genetic variation further contributes to interindividual differences in CYP-mediated drug metabolism. Polymorphisms affecting CYP allele function determine an individual's metabolizer phenotype as a poor, intermediate, normal (extensive), or ultrarapid metabolizer, and thereby influence drug response, efficacy, and toxicity. As pharmacogenomics advances, CYP genotyping is anticipated to play an increasingly prominent role in guiding personalized therapeutic strategies and dose optimization. Such approaches offer the potential to predict individual drug responses more accurately and reduce the incidence of adverse effects.
A representative selection of common CYP inducers, inhibitors, and substrates for major isoforms is summarized below:
CYP1A2
- Inhibitors: Amiodarone, cimetidine, ciprofloxacin, and fluvoxamine
- Inducers: Carbamazepine, phenobarbital, rifampin, and tobacco
- Substrates: caffeine, clozapine, and theophylline
CYP2C9
- Inhibitors: Amiodarone, fluconazole, fluoxetine, metronidazole, ritonavir, and trimethoprim/sulfamethoxazole
- Inducers: Carbamazepine, phenobarbital, phenytoin, and rifampin
- Substrates: carvedilol, celecoxib, glipizide, ibuprofen, irbesartan, losartan, and cannabinoids
CYP2C19
- Inhibitors: Fluvoxamine, isoniazid, and ritonavir
- Inducers: Carbamazepine, phenytoin, and rifampin
- Substrates: Omeprazole and phenobarbital
CYP2D6
- Inhibitors: Bupropion, duloxetine, fluoxetine, paroxetine, quinidine, ritonavir, sertraline, and terbinafine
- Inducers: None
- Substrates: Amitriptyline, carvedilol, codeine, dextromethorphan, diltiazem, donepezil, haloperidol, metoprolol, nifedipine, ondansetron, oxycodone, propranolol, risperidone, tamoxifen, and tramadol
CYP2E1
- Inhibitors: None
- Inducers: Ethanol, isoniazid, and tobacco
- Substrates: Acetaminophen, theophylline, and verapamil
CYP3A4
- Inhibitors: Amiodarone, amitriptyline, aprepitant, carvedilol, chloramphenicol, cimetidine, ciprofloxacin, clarithromycin, codeine, donepezil, fluvoxamine, haloperidol, imatinib, ketoconazole, metoprolol, paroxetine, risperidone, ritonavir, tramadol, and verapamil
- Inducers: Carbamazepine, griseofulvin, phenobarbital, phenytoin, rifampin, and St. John's wort
- Substrates: Alprazolam, amlodipine, buspirone, calcium channel blockers, caffeine, citalopram, clopidogrel, cocaine, cyclosporine, diazepam, erythromycin, estradiol, lidocaine, losartan, many chemotherapeutic drugs, montelukast, quetiapine, sertraline, sildenafil, statins, tacrolimus, warfarin, and zolpidem
Many agents act simultaneously as substrates, inducers, or inhibitors of a given isoform; to avoid redundancy, each medication is listed only once. In addition to their well-recognized role in xenobiotic metabolism, CYP enzymes participate in biological processes relevant to oncology. Several CYP isoforms involved in the metabolism of antitumor drugs belong to the CYP1, CYP2, and CYP3 families. CYP1B1, in particular, has been implicated in tumor progression and chemoresistance and has emerged as a potential therapeutic target. Other isoforms under investigation for cancer applications include CYP2J2 in breast cancer and CYP2W1 in colorectal cancer.[18]
CYP enzymes also serve as clinically actionable targets in endocrine and metabolic diseases. Several Food and Drug Administration–approved therapeutics inhibit steroidogenic CYP isoforms: aromatase inhibitors (letrozole, anastrozole, exemestane) for estrogen-receptor–positive breast cancer; CYP17A1 inhibitors (abiraterone) for prostate cancer; and agents such as osilodrostat and levoketoconazole for Cushing disease. Additional drugs that target CYP5A1 to inhibit thromboxane production and exert antiplatelet effects include picotamide, riogrel, ozagrel, and furegrelate.[13] Moreover, azole antifungals exert their therapeutic activity by inhibiting fungal CYP450 enzymes essential for ergosterol synthesis.[19][20][21]
Media
(Click Image to Enlarge)
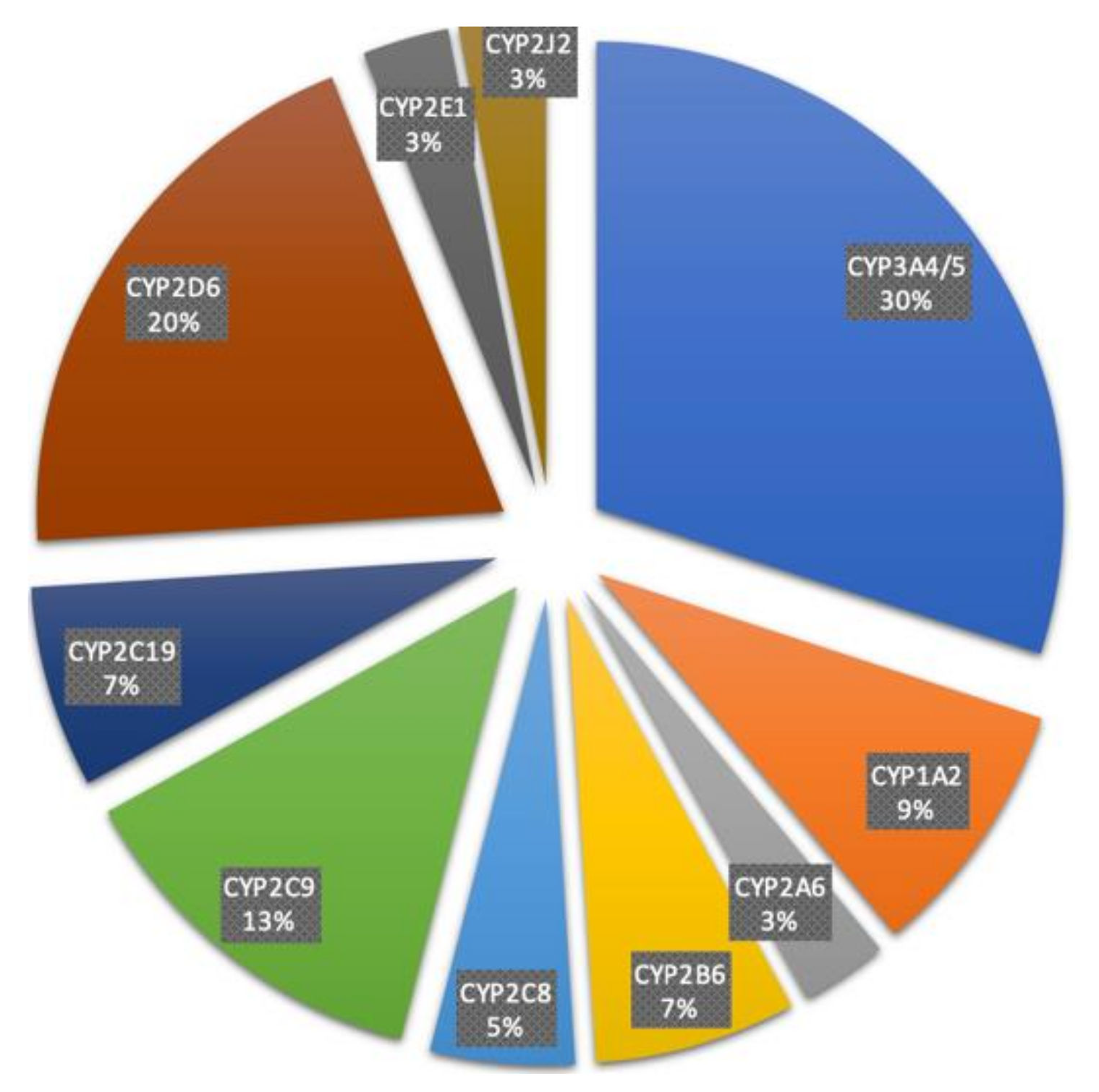
Fractional Contribution of Specific CYP Isoforms to the Metabolism of 248 Drugs.
Zhao M, Ma J, Li M, et al. Cytochrome P450 enzymes and drug metabolism in humans. Int J Mol Sci. 2021;22(23):12808. doi: 10.3390/ijms222312808.
(Click Image to Enlarge)
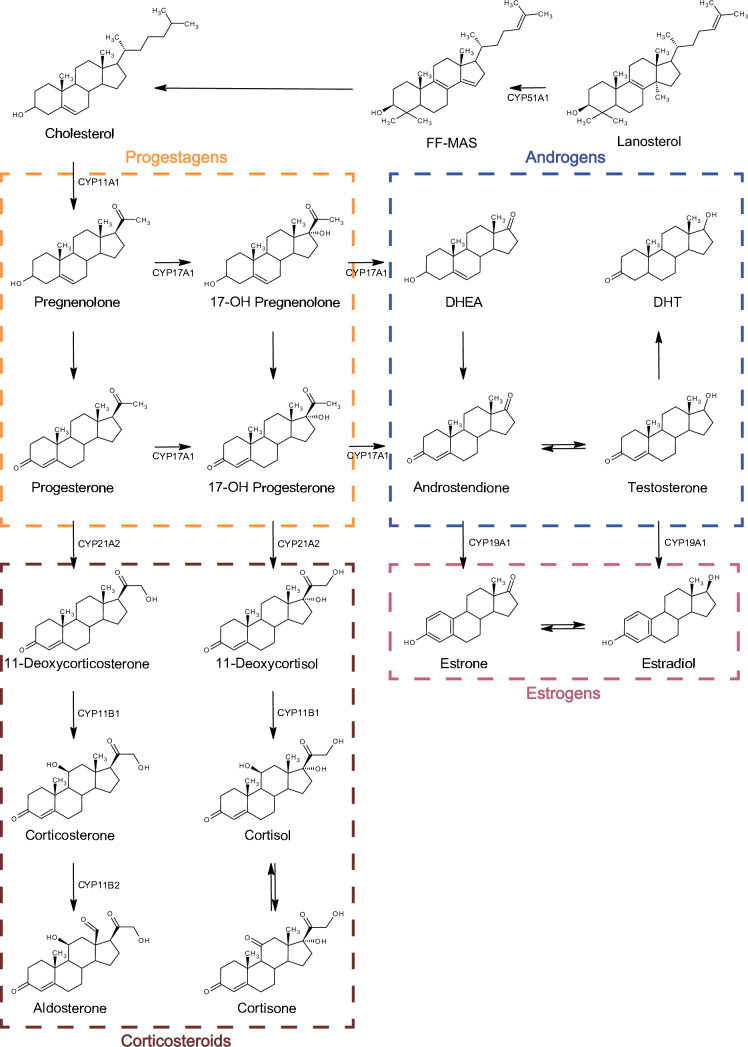
Steroid biosynthesis pathway. FF-MAS (follicular fluid meiosis-activating sterol, 14-demethyl-14-dehydrolanosterol), DHEA (dehydroepiandrosterone), and DHT (dihydrotestosterone)
Toxicol Res. 2024 Apr 22;40(3):325–333. doi: 10.1007/s43188-024-00237-0
References
Rendic S, Guengerich FP. Survey of Human Oxidoreductases and Cytochrome P450 Enzymes Involved in the Metabolism of Xenobiotic and Natural Chemicals. Chemical research in toxicology. 2015 Jan 20:28(1):38-42. doi: 10.1021/tx500444e. Epub 2014 Dec 19 [PubMed PMID: 25485457]
Level 3 (low-level) evidenceBernhardt R. Cytochromes P450 as versatile biocatalysts. Journal of biotechnology. 2006 Jun 25:124(1):128-45 [PubMed PMID: 16516322]
Level 3 (low-level) evidenceStavropoulou E, Pircalabioru GG, Bezirtzoglou E. The Role of Cytochromes P450 in Infection. Frontiers in immunology. 2018:9():89. doi: 10.3389/fimmu.2018.00089. Epub 2018 Jan 31 [PubMed PMID: 29445375]
Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet (London, England). 2002 Oct 12:360(9340):1155-62 [PubMed PMID: 12387968]
Level 3 (low-level) evidenceZahoor I, Rui B, Khan J, Datta I, Giri S. An emerging potential of metabolomics in multiple sclerosis: a comprehensive overview. Cellular and molecular life sciences : CMLS. 2021 Apr:78(7):3181-3203. doi: 10.1007/s00018-020-03733-2. Epub 2021 Jan 15 [PubMed PMID: 33449145]
Level 3 (low-level) evidenceMcDonnell AM, Dang CH. Basic review of the cytochrome p450 system. Journal of the advanced practitioner in oncology. 2013 Jul:4(4):263-8 [PubMed PMID: 25032007]
Ung YT, Ong CE, Pan Y. Current High-Throughput Approaches of Screening Modulatory Effects of Xenobiotics on Cytochrome P450 (CYP) Enzymes. High-throughput. 2018 Sep 29:7(4):. doi: 10.3390/ht7040029. Epub 2018 Sep 29 [PubMed PMID: 30274310]
Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & therapeutics. 2013 Apr:138(1):103-41. doi: 10.1016/j.pharmthera.2012.12.007. Epub 2013 Jan 16 [PubMed PMID: 23333322]
Level 3 (low-level) evidenceManikandan P, Nagini S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Current drug targets. 2018:19(1):38-54. doi: 10.2174/1389450118666170125144557. Epub [PubMed PMID: 28124606]
Elfaki I, Mir R, Almutairi FM, Duhier FMA. Cytochrome P450: Polymorphisms and Roles in Cancer, Diabetes and Atherosclerosis. Asian Pacific journal of cancer prevention : APJCP. 2018 Aug 24:19(8):2057-2070 [PubMed PMID: 30139042]
Almazroo OA, Miah MK, Venkataramanan R. Drug Metabolism in the Liver. Clinics in liver disease. 2017 Feb:21(1):1-20. doi: 10.1016/j.cld.2016.08.001. Epub 2016 Oct 15 [PubMed PMID: 27842765]
Zhao M, Ma J, Li M, Zhang Y, Jiang B, Zhao X, Huai C, Shen L, Zhang N, He L, Qin S. Cytochrome P450 Enzymes and Drug Metabolism in Humans. International journal of molecular sciences. 2021 Nov 26:22(23):. doi: 10.3390/ijms222312808. Epub 2021 Nov 26 [PubMed PMID: 34884615]
Level 2 (mid-level) evidenceGuengerich FP. Roles of cytochrome P450 enzymes in pharmacology and toxicology: Past, present, and future. Advances in pharmacology (San Diego, Calif.). 2022:95():1-47. doi: 10.1016/bs.apha.2021.12.001. Epub 2022 Jul 18 [PubMed PMID: 35953152]
Level 3 (low-level) evidenceBurris-Hiday SD, Scott EE. Allosteric modulation of cytochrome P450 enzymes by the NADPH cytochrome P450 reductase FMN-containing domain. The Journal of biological chemistry. 2023 Sep:299(9):105112. doi: 10.1016/j.jbc.2023.105112. Epub 2023 Jul 28 [PubMed PMID: 37517692]
Guengerich FP. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS catalysis. 2018 Dec 7:8(12):10964-10976. doi: 10.1021/acscatal.8b03401. Epub 2018 Oct 18 [PubMed PMID: 31105987]
Baker HW, Burger HG, de Kretser DM, Dulmanis A, Hudson B, O'Connor S, Paulsen CA, Purcell N, Rennie GC, Seah CS, Taft HP, Wang C. A study of the endocrine manifestations of hepatic cirrhosis. The Quarterly journal of medicine. 1976 Jan:45(177):145-78 [PubMed PMID: 769039]
Bastien MC, Leblond F, Pichette V, Villeneuve JP. Differential alteration of cytochrome P450 isoenzymes in two experimental models of cirrhosis. Canadian journal of physiology and pharmacology. 2000 Nov:78(11):912-9 [PubMed PMID: 11100940]
Level 3 (low-level) evidenceLynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. American family physician. 2007 Aug 1:76(3):391-6 [PubMed PMID: 17708140]
Liu J, Lu YF, Corton JC, Klaassen CD. Expression of cytochrome P450 isozyme transcripts and activities in human livers. Xenobiotica; the fate of foreign compounds in biological systems. 2021 Mar:51(3):279-286. doi: 10.1080/00498254.2020.1867929. Epub 2020 Dec 28 [PubMed PMID: 33350342]
Wang Y, He X, Li C, Ma Y, Xue W, Hu B, Wang J, Zhang T, Zhang F. Carvedilol serves as a novel CYP1B1 inhibitor, a systematic drug repurposing approach through structure-based virtual screening and experimental verification. European journal of medicinal chemistry. 2020 May 1:193():112235. doi: 10.1016/j.ejmech.2020.112235. Epub 2020 Mar 16 [PubMed PMID: 32203789]
Level 1 (high-level) evidenceKim C, Jeong E, Lee YB, Kim D. Steroidogenic cytochrome P450 enzymes as drug target. Toxicological research. 2024 Jul:40(3):325-333. doi: 10.1007/s43188-024-00237-0. Epub 2024 Apr 22 [PubMed PMID: 38911541]