Introduction
Congenital pulmonary airway malformation (CPAM), previously known as congenital cystic adenomatoid malformation, falls within the broader category of congenital lung malformations.[1] Although rare, CPAM represents the most common form of congenital lung malformation. Other congenital lung malformations requiring differentiation from CPAM include bronchopulmonary sequestration, bronchogenic cyst, congenital lobar emphysema, and related developmental pulmonary anomalies.[2] Please see StatPearls' companion resources, "Pulmonary Sequestration," "Bronchogenic Cyst," and "Congenital Lobar Emphysema," for further information.
Abnormalities during embryogenesis and fetal lung development lead to anomalous bronchial morphogenesis and subsequent formation of CPAM lesions. Advances in fetal imaging and prenatal diagnostic technologies have increased the frequency of antenatal detection, allowing earlier identification and risk assessment of affected fetuses. Clinical presentation demonstrates substantial variability, ranging from asymptomatic patients who remain undiagnosed until later childhood or adulthood to severe neonatal respiratory distress and lesions incompatible with life.[2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of CPAM remains multifactorial, involving complex disruptions in normal pulmonary development. CPAM arises from abnormalities in lung formation across the 5 stages of embryogenesis, including the embryonic, pseudoglandular, canalicular, saccular, and alveolar phases, each contributing to the structural and functional maturation of the respiratory system.
Multiple genes involved in early lung development have been implicated in CPAM pathogenesis. These include the thyroid transcription factor gene (Nkx2), sex-determining region Y-box 2 gene (Sox2), Hox gene (Hoxb-5), Ying Yang 1 gene (Yy1), fatty acid-binding protein-7 gene (FABP-7), acyl-CoA synthetase 5 (ACSL5), platelet-derived growth factor B gene (PDGF-B), sonic hedgehog (SHH), bone morphogenetic protein 4 (BMP4), sprouty 2 (SPRY2), Wnt2B, transforming growth factor beta (TGFB), and fibroblast growth factors 10, 9, and 7 (FGF10, FGF9, FGF7). These genes regulate key processes, eg, cellular proliferation, differentiation, and apoptosis, and abnormal expression patterns may contribute to the development of congenital lung malformations, including CPAM.[2][3][4] Additional research has also explored a potential association with KRAS mutations, suggesting a possible role for oncogenic signaling pathways in lesion formation.[5]
Epidemiology
Although rare, CPAM accounts for about 95% of congenital cystic lung disease. The incidence of CPAM is reported to range from 1 in 10,000 to 1 in 35,000 births.[3]
Pathophysiology
CPAM is differentiated into 5 major types by the Stocker classification. Each type originates from a different part of the bronchial tree, resulting in distinct histopathological features, clinical manifestations, malignant potential, and overall prognosis.[1][2][6][7] CPAM is mostly unilateral, involving 1 lobe, and has an intact blood supply from the pulmonary arteries, and maintains communication with the tracheopulmonary tree.[1][6] The Stocker classification divides CPAM into 5 types based on the following features:
- Type 0: This type is rare (1%-3%) and originates in the trachea or bronchi with histology showing bronchial-type airways with cartilage, smooth muscles, and glands separated by mesenchyme. Type 0 involves both lungs as a diffuse malformation, leading to no gas exchange and therefore is considered lethal or incompatible with life.
- Type 1: This type accounts for the majority of CPAM cases (50%-70%) and originates in the bronchi. The cysts in this type are usually single or multiloculated, located primarily within 1 lobe, and have a lining of pseudo-stratified columnar epithelium. Type 1 has a good prognosis following resection and rarely undergoes malignant transformation (bronchiolo-alveolar carcinoma). The clinical presentation depends on the size of the cysts and could lead to respiratory distress after birth.
- Type 2: This is the second most frequent type (10%-40%), originating in the bronchiolar regions and often associated with bronchial atresia and other anomalies (renal agenesis, cardiovascular defects, diaphragmatic hernia, skeletal defects, esophageal atresia). Usually, type 2 presents as multiple small cysts with no mass effect. The prognosis is variable and has no malignant potential.
- Type 3: This type is less common (5%-10%) and originates in the alveolar regions. Type 3 typically involves an entire lobe, causing compression of the other lobes. These lesions are usually solid versus cystic. The prognosis is poor, as type 3 CPAM can cause fetal hydrops with lung hypoplasia and can present as severe respiratory distress at birth. Furthermore, this type has no malignant potential.
- Type 4: This type is also less common (10%-15%) and originates in the acinar structures of the lung. Type 4 consists of single or multifocal peripheral thin-walled cysts filled with air and can present as a pneumothorax. Type 4 CPAM has a high malignant potential, similar to that of type 1 pleuropulmonary blastomas. Prognosis can be good with timely surgical intervention.
History and Physical
CPAM demonstrates a broad spectrum of clinical presentations (see Table 1). At one extreme, CPAM may present with severe disease incompatible with life or manifest as significant respiratory distress immediately after birth. At the opposite end of the spectrum, affected infants may remain completely asymptomatic throughout infancy and into childhood, with no overt clinical manifestations.
Advances in fetal imaging, particularly the widespread use of prenatal ultrasonography for the evaluation of fetal lung abnormalities, have increased the rate of antenatal diagnosis. This shift toward earlier detection has contributed to a relative decrease in the proportion of symptomatic CPAM cases at birth. Despite the absence of early symptoms, asymptomatic neonates remain at risk for later complications during childhood, including recurrent respiratory infections and rare malignant transformation. Symptomatic neonates typically present with respiratory distress, with severity closely related to lesion size and the degree of compression on adjacent airways and mediastinal structures. Each CPAM subtype demonstrates distinct clinical and radiologic characteristics that further define presentation patterns and prognostic implications.[8][9]
Table 1. Clinical Presentations of Congenital Pulmonary Airway Malformation
| Type of CPAM | Common Clinical Presentation |
| Type 0 | Incompatible with life/lethal |
| Type 1 | Respiratory distress, tachypnea, and cyanosis. |
| Type 2 | Respiratory distress, with additional congenital anomalies (renal agenesis, cardiovascular defects, diaphragmatic hernia) |
| Type 3 | Nonimmune fetal hydrops with pulmonary hypoplasia presenting as severe respiratory distress after birth. |
| Type 4 | Pneumothorax leading to respiratory distress, small risk of infection, malignancy, air leak, or bleeding |
Evaluation
CPAM is the most common lung malformation diagnosed prenatally. A second-trimester fetal ultrasound is the primary imaging modality for detecting CPAM, as they appear more echogenic than the rest of the lung tissue, and can be classified as either microcystic or macrocystic based on the size.[1] For further characterization of the lesion, fetal magnetic resonance imaging (MRI) is recommended due to its high sensitivity and specificity for delineating the type and extent of lung lesions, especially in more complex cases.[3]
A significant prognostic indicator is the CPAM Volume Ratio (CVR), which is calculated by dividing the CPAM lesion volume by the head circumference at that gestational age. A CVR of more than 1.6 is strongly associated with fetal hydrops and inferior clinical outcomes, with few studies suggesting a CVR of greater than 2 as a more critical threshold.[10] CVR value helps guide early referral to a high-risk fetal center. A fetal echocardiogram should be performed to evaluate cardiac function in severe cases.[2]
If an infant has a diagnosis of CPAM from a prenatal ultrasound and is symptomatic after birth, an initial chest radiograph is usually performed as a part of management.[11] For diagnostic imaging, a computed tomography (CT) scan is considered the gold standard. Postnatal MRI can be performed, but its accuracy is limited, and evidence of its superiority over the CT scan is lacking.[3][10] In asymptomatic infants, CT of the lungs is recommended by 3 months of age.[3] In infants with the absence of a prenatal diagnosis of CPAM due to no prenatal imaging, infection, eg, pneumonia, requiring a chest x-ray often leads to the diagnosis of CPAM, while in many cases, CPAM is detected as an incidental finding in a postnatal chest x-ray showing hyperlucency with a possible radio-opaque component.[2]
Treatment / Management
Management of congenital pulmonary airway malformation can be performed in the prenatal and postnatal period, depending on the severity and presentation (see Table 2).[6] In the prenatal period, if the risk of nonimmune fetal hydrops is identified, interventions, eg, maternal corticosteroids (betamethasone) are indicated to potentially reduce CPAM size and improve hydrops.[10] For cases with worsening hydrops, procedures like cyst drainage, thoracoamniotic shunt, or open resection can be considered to improve prenatal outcomes.[6] In selected cases with large lung masses and mediastinal shift, ex utero intrapartum (EXIT) procedures can be performed at specialized centers.[2] The presence of a high CVR ratio, fetal hydrops, and mediastinal shift can guide delivery at a higher-level center with adequate facilities (eg, pediatric surgery) and extracorporeal membrane oxygenation (ECMO), to manage affected newborns.[6][10] (B3)
In infants presenting with symptomatic CPAM after birth, immediate respiratory support within the neonatal intensive care unit remains essential to stabilize respiratory function and address hypoxemia or respiratory distress. Surgical resection represents the definitive management strategy for symptomatic cases.[6] Lobectomy constitutes the most frequently performed operative approach, although lung-sparing procedures such as segmentectomy may be appropriate in selected cases to preserve functional pulmonary tissue while ensuring complete lesion removal.[1][10](B3)
In asymptomatic patients, no universally preferred strategy exists for determining the optimal timing of surgical intervention. Early elective resection, commonly performed between 6 and 12 months of age, offers potential benefits including enhanced compensatory lung growth, reduction in infection risk, mitigation of potential malignant transformation, and decreased cumulative exposure to diagnostic radiation. Alternatively, a conservative management strategy emphasizes close clinical and radiologic surveillance, with delayed surgical intervention reserved for patients who develop symptoms or complications. This approach considers the possibility of spontaneous lesion regression, the generally low malignant potential in many CPAM cases, and the feasibility of timely intervention if clinical deterioration occurs. Management decisions require individualized assessment by the multidisciplinary care team in collaboration with the patient’s family, weighing risks and benefits of both operative and nonoperative strategies.[6][10][12][13] Following surgical resection or during conservative management of stable asymptomatic CPAM, longitudinal follow-up with periodic computed tomography imaging supports ongoing surveillance for recurrence, residual disease, or late-onset complications, with imaging intervals tailored to clinical status and risk profile.[3](B2)
Table 2. Congenital Pulmonary Airway Malformation Prenatal Diagnosis and Management
|
Imaging Modality |
|
|
Postdiagnostic Evaluation |
Assessment for associated congenital anomalies is present in 10%–20% of CPAMs
|
|
Differential Diagnosis |
|
|
Prenatal Course |
Most CPAMs demonstrate rapid, progressive growth from 20 to 26 weeks, peaking at approximately 25 weeks, then plateau and often regress. If hydrops has not developed by 28 weeks, it is unlikely to develop, and the prognosis is excellent. |
|
Poor Prognostic Factors |
|
|
Management of Hydrops |
|
|
Delivery |
|
Differential Diagnosis
The primary differential diagnoses for CPAM include other congenital lung malformations (CLMs), particularly bronchopulmonary sequestration, bronchogenic cyst, and congenital lobar emphysema.[2] Bronchopulmonary sequestration can also be detected prenatally by ultrasonography and typically appears as a well-defined, solid, homogeneous mass. These lesions remain nonfunctional and, unlike CPAM, lack communication with the tracheobronchial tree. Their vascular supply originates from the systemic circulation through an anomalous systemic artery rather than from the pulmonary circulation.
Bronchogenic cyst, commonly referred to as a foregut duplication cyst, presents as a fluid-filled cystic lesion without communication with the tracheobronchial tree. Congenital lobar emphysema, also termed congenital lobar overinflation, causes hyperinflation of a pulmonary lobe and commonly appears hyperlucent on chest radiography with increased echogenicity on prenatal ultrasonography.[1][6] Additional prenatal differential diagnoses include congenital diaphragmatic hernia and other fetal thoracic masses, all of which require careful imaging evaluation to establish an accurate diagnosis and guide appropriate perinatal management.[14]
Prognosis
The overall prognosis of CPAM varies considerably and largely depends on the specific CPAM subtype. Several reported cases have demonstrated prenatal regression of the lesion, particularly with close fetal monitoring and appropriate perinatal management. The presence of fetal hydrops significantly worsens outcomes and correlates with decreased survival rates. Surgical resection during the neonatal period has been shown to improve survival and often provides definitive treatment. Studies have also suggested that, in carefully selected patients, conservative management may achieve outcomes comparable to surgical intervention.[13]
Among the CPAM subtypes classified by the Stocker system, type 1 CPAM carries the most favorable prognosis. The prognosis of type 2 CPAM depends largely on the severity and extent of associated congenital anomalies. Type 3 CPAM commonly involves hypoplasia of an entire lobe, which can contribute to serious complications, eg, pulmonary hypertension due to compromised lung development and mass effect on adjacent structures. Type 4 CPAM generally demonstrates an excellent prognosis following surgical resection; however, this subtype maintains a strong association with pleuropulmonary blastoma, necessitating careful long-term surveillance and clinical follow-up.[1][2][6]
Complications
Infection, particularly pneumonia, and development of pneumothorax represent common complications of CPAM and most frequently occur during the first few years of life, especially when conservative management delays surgical resection. Patients managed nonoperatively require close clinical surveillance because recurrent respiratory infections and progressive pulmonary complications may develop over time. Following surgical resection, potential postoperative complications include residual lesions, infection, bleeding, spontaneous pneumothorax, persistent air leak, and other procedure-related pulmonary complications.[15]
Pleuropulmonary blastoma (PPB) remains the most common malignant complication associated with CPAM. Risk factors for PPB include CPAM type 4, multifocal or bilateral cystic lesions, and a family history of associated tumors.[11][15] CPAM type 1 rarely undergoes malignant transformation into bronchioloalveolar carcinoma. Pulmonary hypoplasia with subsequent pulmonary hypertension may occur in CPAM type 3 because the lesion can involve an entire lobe or even the entire lung.[6] One study suggested an increased risk for reduced lung function and diminished exercise tolerance in children with CPAM requiring surgical intervention.[16] Another study demonstrated no significant difference in long-term respiratory function between infants undergoing surgery before or after 2 years of age.[17]
Deterrence and Patient Education
Deterrence and patient education in CPAM focus on early recognition, timely surveillance, prevention of complications, understanding the disease's prognosis, natural history, and complications, and informed family participation in management decisions. Although no established preventive measures exist because CPAM develops during embryogenesis, prenatal detection through routine fetal ultrasonography allows clinicians to identify high-risk lesions and coordinate early referral to specialized fetal care centers. Families should receive education regarding the variable clinical spectrum of CPAM, including the possibility of asymptomatic disease, neonatal respiratory distress, recurrent pulmonary infections, pneumothorax, pulmonary hypoplasia, and rare malignant transformation. Counseling should also address the importance of serial prenatal imaging, postnatal CT evaluation, and adherence to scheduled follow-up appointments to detect lesion progression or complications that require intervention.
Patient and caregiver education should emphasize recognition of respiratory symptoms such as tachypnea, wheezing, recurrent pneumonia, feeding difficulty, cyanosis, or exercise intolerance that warrant prompt medical evaluation. Clinicians should discuss the benefits and limitations of conservative monitoring versus elective surgical resection, including the risks of infection, malignancy, and postoperative complications. Shared decision-making between healthcare professionals and families improves adherence to monitoring strategies and follow-up appointments while reducing delays in treatment. Education provided by physicians, advanced practitioners, nurses, respiratory therapists, and pharmacists supports safe postoperative recovery, medication adherence, and long-term pulmonary health surveillance, ultimately improving patient-centered outcomes and quality of care.
Enhancing Healthcare Team Outcomes
CPAM is the most common congenital lung malformation and results from abnormal embryologic development of the tracheobronchial tree during fetal lung formation. CPAM demonstrates variable clinical severity ranging from asymptomatic lesions identified prenatally to life-threatening neonatal respiratory distress, pulmonary hypoplasia, fetal hydrops, or mediastinal compression. The Stocker classification categorizes CPAM into 5 subtypes with distinct histopathologic features, prognostic implications, and malignant potential. Prenatal ultrasonography and fetal MRI play central roles in diagnosis, lesion characterization, and risk assessment, while postnatal CT imaging remains the diagnostic gold standard. Management depends on lesion size, symptom severity, and associated complications and may include maternal corticosteroids, fetal intervention, neonatal respiratory support, surgical resection, or conservative monitoring. Long-term complications include recurrent infection, pneumothorax, pulmonary hypertension, impaired lung function, and rare malignant transformation such as pleuropulmonary blastoma or bronchioloalveolar carcinoma.
Interprofessional collaboration improves outcomes through coordinated prenatal assessment, delivery planning, neonatal stabilization, surgical decision-making, and longitudinal follow-up. Maternal-fetal medicine specialists, neonatologists, pediatric surgeons, radiologists, pulmonologists, primary care clinicians, advanced practitioners, nurses, respiratory therapists, and pharmacists contribute to timely diagnosis, risk stratification, and complication prevention. Radiologists interpret fetal and postnatal imaging to guide prognosis and referral, while pediatric surgeons evaluate indications for resection and postoperative monitoring. Nurses and respiratory therapists provide respiratory assessment, ventilatory support, family education, and postoperative care. Pharmacists support safe medication management, including corticosteroid therapy and perioperative treatment. Primary care clinicians coordinate surveillance, monitor growth and respiratory status, reinforce follow-up adherence, and facilitate communication among specialists. Shared decision-making with families regarding surgical versus conservative management supports patient-centered care, improves adherence, and reduces preventable morbidity through early recognition of complications and timely referral to specialized centers.
Media
(Click Image to Enlarge)
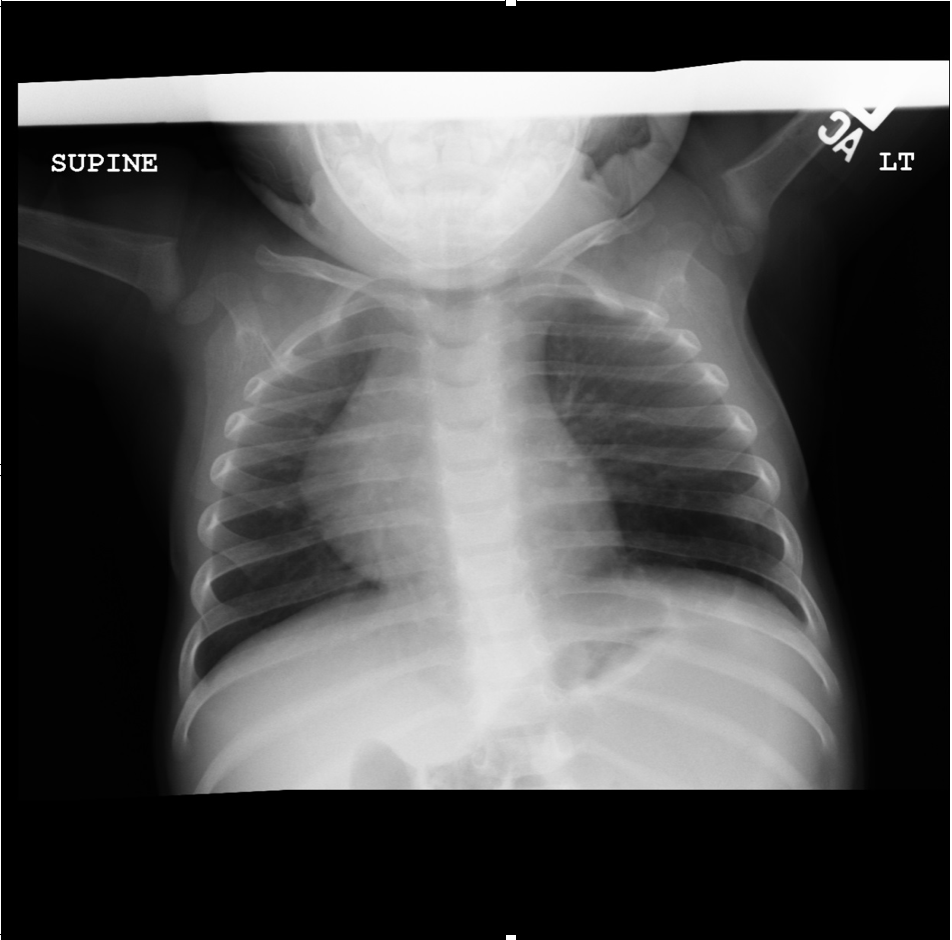
AP view of chest radiograph showing left lower lobe CPAM Courtesy James Cameron, MD, Rush University Medical Center, Chicago, USA
(Click Image to Enlarge)
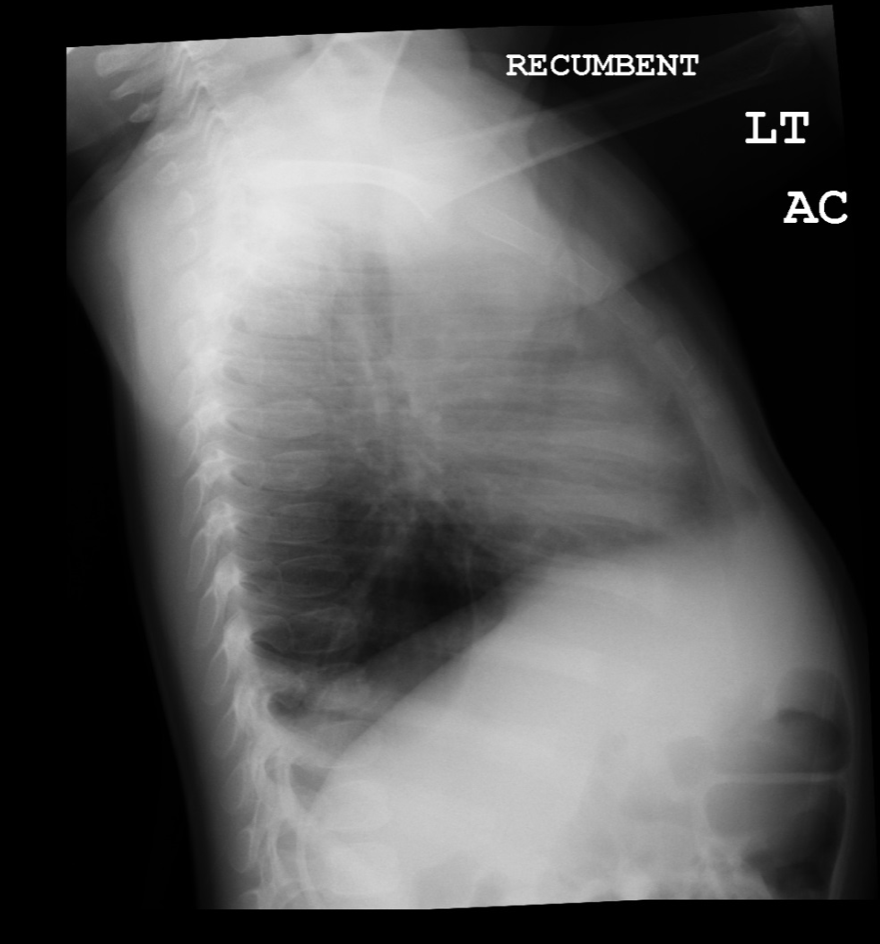
Lateral view of chest radiograph showing left lower lobe CPAM Courtesy James Cameron, MD, Rush University Medical Center, Chicago, USA
(Click Image to Enlarge)
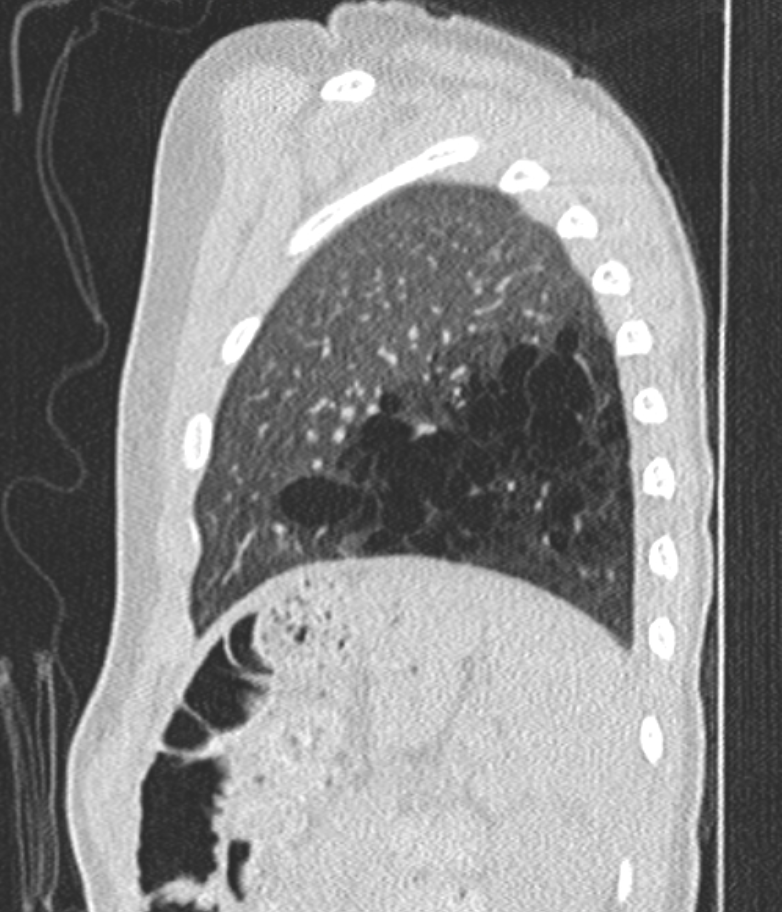
CT chest image (sagittal view) showing left lower lobe CPAM Courtesy James Cameron, MD, Rush University Medical Center, Chicago, USA
(Click Image to Enlarge)
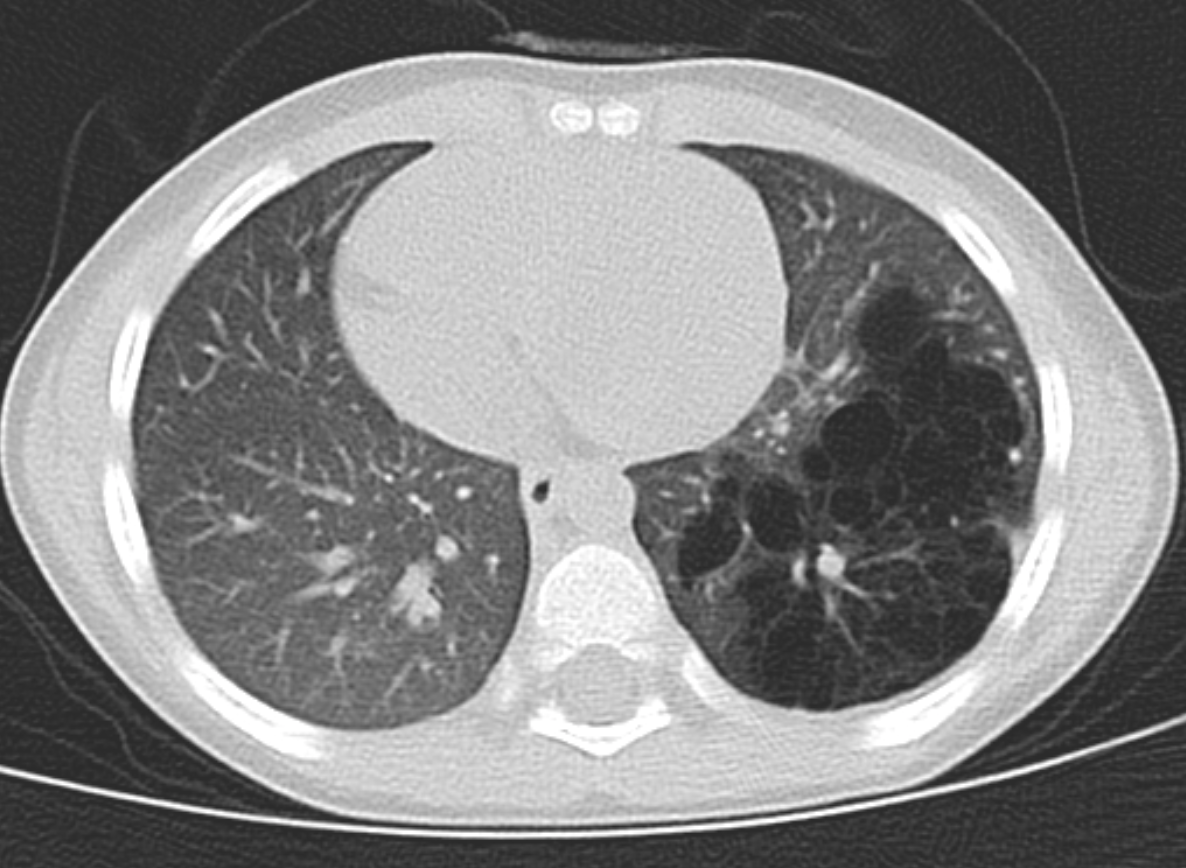
CT chest (coronal view) showing left lower lobe CPAM Courtesy James Cameron, MD, Rush University Medical Center, Chicago, USA
(Click Image to Enlarge)
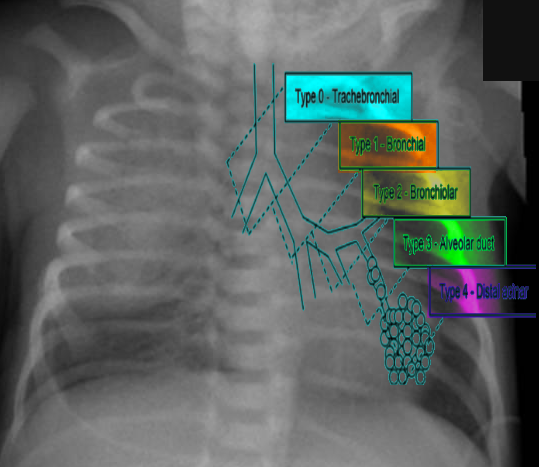
Types of CPAM Image courtesy S Bhimji
References
Hegde BN, Tsao K, Hirose S. Management of Congenital Lung Malformations. Clinics in perinatology. 2022 Dec:49(4):907-926. doi: 10.1016/j.clp.2022.08.003. Epub [PubMed PMID: 36328607]
Leblanc C, Baron M, Desselas E, Phan MH, Rybak A, Thouvenin G, Lauby C, Irtan S. Congenital pulmonary airway malformations: state-of-the-art review for pediatrician's use. European journal of pediatrics. 2017 Dec:176(12):1559-1571. doi: 10.1007/s00431-017-3032-7. Epub 2017 Oct 19 [PubMed PMID: 29046943]
David M, Lamas-Pinheiro R, Henriques-Coelho T. Prenatal and Postnatal Management of Congenital Pulmonary Airway Malformation. Neonatology. 2016:110(2):101-15. doi: 10.1159/000440894. Epub 2016 Apr 13 [PubMed PMID: 27070354]
Qu Y, Liu D, Jia H, Zhou X. Expression Analysis of ACSL5 and Wnt2B in Human Congenital Pulmonary Airway Malformations. The Journal of surgical research. 2018 Dec:232():128-136. doi: 10.1016/j.jss.2018.06.023. Epub 2018 Jul 4 [PubMed PMID: 30463708]
Nelson ND, Xu F, Chandrasekaran P, Litzky LA, Peranteau WH, Frank DB, Li M, Pogoriler J. Defining the spatial landscape of KRAS mutated congenital pulmonary airway malformations: a distinct entity with a spectrum of histopathologic features. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2022 Dec:35(12):1870-1881. doi: 10.1038/s41379-022-01129-0. Epub 2022 Jul 6 [PubMed PMID: 35794233]
Dias JF, Dias MB, Rocha G. Congenital Lung Malformations: A Comprehensive Overview of Current Knowledge-Narrative Review. Archivos de bronconeumologia. 2026 Feb:62(2):104-112. doi: 10.1016/j.arbres.2025.09.021. Epub 2025 Oct 4 [PubMed PMID: 41109832]
Level 3 (low-level) evidenceFowler DJ, Gould SJ. The pathology of congenital lung lesions. Seminars in pediatric surgery. 2015 Aug:24(4):176-82. doi: 10.1053/j.sempedsurg.2015.02.002. Epub 2015 Feb 27 [PubMed PMID: 26051050]
Maneenil G, Ruangnapa K, Thatrimontrichai A, Janjindamai W, Dissaneevate S, Anantaseree W, Suntornlohanakul S. Clinical presentation and outcome in congenital pulmonary malformation: 25 year retrospective study in Thailand. Pediatrics international : official journal of the Japan Pediatric Society. 2019 Aug:61(8):812-816. doi: 10.1111/ped.13934. Epub 2019 Jul 2 [PubMed PMID: 31264305]
Level 2 (mid-level) evidenceParikh DH, Rasiah SV. Congenital lung lesions: Postnatal management and outcome. Seminars in pediatric surgery. 2015 Aug:24(4):160-7. doi: 10.1053/j.sempedsurg.2015.01.013. Epub 2015 Feb 3 [PubMed PMID: 26051048]
Baird R, Puligandla PS, Laberge JM. Congenital lung malformations: informing best practice. Seminars in pediatric surgery. 2014 Oct:23(5):270-7. doi: 10.1053/j.sempedsurg.2014.09.007. Epub 2014 Sep 4 [PubMed PMID: 25459011]
Ng C, Stanwell J, Burge DM, Stanton MP. Conservative management of antenatally diagnosed cystic lung malformations. Archives of disease in childhood. 2014 May:99(5):432-7. doi: 10.1136/archdischild-2013-304048. Epub 2014 Jan 9 [PubMed PMID: 24406806]
Muller CO, Berrebi D, Kheniche A, Bonnard A. Is radical lobectomy required in congenital cystic adenomatoid malformation? Journal of pediatric surgery. 2012 Apr:47(4):642-5. doi: 10.1016/j.jpedsurg.2011.08.002. Epub [PubMed PMID: 22498375]
Level 2 (mid-level) evidenceCook J, Chitty LS, De Coppi P, Ashworth M, Wallis C. The natural history of prenatally diagnosed congenital cystic lung lesions: long-term follow-up of 119 cases. Archives of disease in childhood. 2017 Sep:102(9):798-803. doi: 10.1136/archdischild-2016-311233. Epub 2017 Jun 5 [PubMed PMID: 28584070]
Level 3 (low-level) evidenceAdzick NS, Harrison MR, Crombleholme TM, Flake AW, Howell LJ. Fetal lung lesions: management and outcome. American journal of obstetrics and gynecology. 1998 Oct:179(4):884-9 [PubMed PMID: 9790364]
Level 2 (mid-level) evidenceWong A, Vieten D, Singh S, Harvey JG, Holland AJ. Long-term outcome of asymptomatic patients with congenital cystic adenomatoid malformation. Pediatric surgery international. 2009 Jun:25(6):479-85. doi: 10.1007/s00383-009-2371-5. Epub 2009 Apr 30 [PubMed PMID: 19404649]
Hijkoop A, van Schoonhoven MM, van Rosmalen J, Tibboel D, van der Cammen-van Zijp MHM, Pijnenburg MW, Cohen-Overbeek TE, Schnater JM, IJsselstijn H. Lung function, exercise tolerance, and physical growth of children with congenital lung malformations at 8 years of age. Pediatric pulmonology. 2019 Aug:54(8):1326-1334. doi: 10.1002/ppul.24345. Epub 2019 Apr 22 [PubMed PMID: 31012287]
Keijzer R, Chiu PP, Ratjen F, Langer JC. Pulmonary function after early vs late lobectomy during childhood: a preliminary study. Journal of pediatric surgery. 2009 May:44(5):893-5. doi: 10.1016/j.jpedsurg.2009.01.021. Epub [PubMed PMID: 19433164]