Introduction
Cerebral amyloid angiopathy (CAA) is a cerebrovascular disorder characterized by the accumulation of amyloid β-peptide within the leptomeninges and small- to medium-sized cerebral blood vessels.[1] Amyloid deposition creates fragile vessels that may result in lobar intracerebral hemorrhages (ICH). CAA may also present with cognitive impairment, incidental microbleeds, hemosiderosis, inflammatory leukoencephalopathy, Alzheimer disease, or transient neurological symptoms.[2] CAA can also occur in specific familial syndromes or sporadically. Diagnosis is based on a combination of clinical, pathological, and radiographic findings. However, a definitive diagnosis requires a postmortem examination of the brain. Currently, no disease-modifying treatments are available. Prognosis depends on the presenting features, with worse outcomes in older patients with large hematomas.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The exact etiology of cerebral amyloid angiopathy remains unclear. CAA is characterized by deposition of congophilic material (amyloid β-peptide) deposits in the leptomeninges and small- to medium-sized cerebral blood vessels. This deposition weakens vessel walls, making them prone to bleeding. CAA can also occur in specific familial syndromes or sporadically.
Familial CAA
Inherited, or familial, CAA is a rare form of the disease with an autosomal dominant inheritance pattern and an earlier age of onset than sporadic CAA. Kindreds with familial CAA have been reported worldwide, each subtype linked to a specific mutation in the amyloid precursor protein gene. Affected individuals typically present between 30 and 70 years of age, compared with 60 to 90 years of age in the sporadic form. In familial CAA, other mutations encode peptides deposited in the brain, including ACys, ATTR, PrPSc, ABri, ADan, and AGel.[3]
Sporadic CAA
In older adults, the factors underlying amyloid β-peptide deposition remain poorly understood. The most important common genetic factor is the apolipoprotein E (APOE) gene, which encodes a lipid-transport protein in the brain. Both the ε2 and ε4 alleles are more prevalent in patients with CAA and CAA-related lobar intracerebral hemorrhage; ε4 is consistently associated with a higher burden of vascular Aβ deposition, whereas ε2 is linked to more severe vasculopathic changes and a greater tendency toward vessel rupture and hemorrhage.[4]
Epidemiology
Cerebral amyloid angiopathy is strongly age-dependent, with the prevalence of moderate to severe CAA increasing with age. Sporadic CAA is uncommon in individuals aged 60 to 65 years and rare in individuals aged 50 years or younger. CAA does not appear to have a higher prevalence in either sex. Hypertension may coexist and contribute to overall hemorrhage risk, but the underlying vasculopathy in CAA is amyloid-mediated rather than classic hypertensive arteriolosclerosis.
History and Physical
Cerebral amyloid angiopathy is usually asymptomatic. However, when symptomatic, the most common clinical manifestation is spontaneous lobar hemorrhage. The location and the size of the hemorrhage largely determine the clinical deficits. Hemorrhages extending toward the ventricles may cause hemiplegia and decreased consciousness, while smaller hemorrhages may cause more focal deficits, headaches, or seizures. Importantly, very small hemorrhages may be asymptomatic. The hemorrhage location often reflects the distribution of amyloid β-peptide, which often favors cortical vessels. Additionally, the hemorrhages are more likely to occur in the posterior brain. The cerebellum is frequently involved and contains vascular amyloid accumulation (primarily in the cerebellar cortex and vermis).
Cognitive impairment is also a common presenting symptom. Three patterns have been described:
- Gradual decline: microhemorrhages, lobar lacunas, microinfarcts, ischemic leukoencephalopathy
- Stepwise decline: recurrent lobar hemorrhages
- Rapidly progressive decline: cerebral amyloid angiopathy–related inflammation
Another characteristic manifestation is transient focal neurological episodes (TFNEs or “amyloid spells”). These episodes typically present as motor, positive or negative sensory, visual, or language symptoms that recur over days to weeks. These events may correspond to cortical subarachnoid hemorrhage or cortical superficial siderosis on MRI and carry an elevated short-term risk of symptomatic lobar ICH. These neurological episodes must be differentiated from transient ischemic attacks because antithrombotic therapy can worsen or precipitate ICH.[5]
Evaluation
The Boston criteria are commonly used to evaluate patients for cerebral amyloid angiopathy. The Boston criteria version 2.0, published in 2022, was developed to incorporate leptomeningeal and white matter imaging features more fully into the in vivo diagnosis of probable and possible CAA (see Table 1). The criteria integrate clinical, radiologic, and, when available, pathologic information and represent an evolution of the original Boston criteria (1995) and the modified Boston criteria (2010). Definitive diagnoses can only be made through postmortem examination of the brain. However, probable and possible CAA diagnoses can be made using imaging or tissue sampling:
Definite CAA
Requires a complete postmortem brain examination demonstrating:
- A compatible clinical presentation: Spontaneous intracerebral hemorrhage, transient focal neurological episodes, convexity subarachnoid hemorrhage, or cognitive impairment/dementia
- Severe CAA with vasculopathy
- Absence of any other diagnostic lesion
Probable CAA With Supporting Pathology
Requires clinical data plus pathological tissue (evacuated hematoma or cortical biopsy) showing:
- Presentation with spontaneous intracerebral hemorrhage, transient focal neurological episodes, convexity subarachnoid hemorrhage, or cognitive impairment/dementia
- Some degree of CAA in the tissue specimen
- Absence of any other diagnostic lesion
Probable CAA (Without Pathological Confirmation)
Pathological confirmation is not required. Criteria are applied in patients aged 50 years or older who present with:
- Spontaneous intracerebral hemorrhage, transient focal neurological episodes, or cognitive impairment/dementia
MRI criteria (T2-weighted) must demonstrate either:
- At least 2 strictly lobar hemorrhagic lesions in any combination of:
- Intracerebral hemorrhage
- Cerebral microbleeds (See Image. Disseminated Cerebral Amyloid Angiopathies)
- Foci of cortical superficial siderosis (multiple distinct foci counted as separate hemorrhagic lesions)
- Convexity subarachnoid hemorrhage (multiple distinct foci counted as separate hemorrhagic lesions)
OR
- One lobar hemorrhagic lesion plus one white-matter feature:
- Severe perivascular spaces in the centrum semiovale (> 20 visible in one hemisphere), or
- White matter hyperintensities in a multispot pattern (> 10 subcortical fluid-attenuated inversion recovery [FLAIR] dots bilaterally)
In addition, all of the following must be true:
- No deep hemorrhagic lesions on T2-weighted MRI*
- No other cause of hemorrhagic lesions*
Hemorrhagic lesions in the cerebellum are not counted as either lobar or deep hemorrhagic lesions.
Possible CAA (Without Pathological Confirmation)
Pathological confirmation is not required. Criteria are applied in patients aged 50 years or older who present with:
- Spontaneous intracerebral hemorrhage, transient focal neurological episodes, or cognitive impairment/dementia
MRI criteria (T2-weighted) must demonstrate either:
- One strictly lobar hemorrhagic lesion, defined as:
- Intracerebral hemorrhage
- Cerebral microbleeds
- Foci of cortical superficial siderosis
- Convexity subarachnoid hemorrhage
OR
- One white matter feature alone, defined as:
- Severe perivascular spaces in the centrum semiovale (> 20 visible in one hemisphere), or
- White matter hyperintensities in a multispot pattern (> 10 subcortical fluid-attenuated inversion recovery FLAIR dots bilaterally)
All of the following must be true:
- No deep hemorrhagic lesions on T2-weighted MRI
- No other cause of hemorrhagic lesions*
Hemorrhagic lesions in the cerebellum are not counted as lobar or deep hemorrhagic lesions.
*Other causes of hemorrhagic lesions include: antecedent head trauma, hemorrhagic transformation of an ischemic stroke, arteriovenous malformation, hemorrhagic tumor, warfarin therapy with an international normalized ratio greater than 3, and vasculitis.
Table 1. Boston Criteria Version 2.0 for Cerebral Amyloid Angiopathy (Summary)[2]
|
Tier |
Age |
Imaging/pathology requirements |
Exclusions |
|
Definite CAA |
Any |
Complete postmortem brain examination demonstrating severe CAA with vasculopathy and no alternative diagnostic lesion |
Requires autopsy |
|
Probable CAA with supporting pathology |
Any |
Clinical presentation compatible with CAA plus histologic evidence of CAA in evacuated hematoma or cortical biopsy, with no other diagnostic lesion |
Requires surgical/biopsy tissue |
|
Probable CAA (no pathology required) |
≥ 50 years |
MRI (T2*/SWI): either (a) ≥2 strictly lobar hemorrhagic lesions (lobar ICH, lobar CMBs, cSS foci, or cSAH; multiple distinct foci counted separately), OR (b) 1 strictly lobar hemorrhagic lesion plus 1 white matter feature (severe CSO-PVS > 20 in one hemisphere, or multispot WMH pattern with > 10 small subcortical FLAIR dots bilaterally) |
No deep hemorrhagic lesions and no other apparent cause of hemorrhagic lesions* |
|
Possible CAA (no pathology required) |
≥ 50 years |
MRI: either (a) 1 strictly lobar hemorrhagic lesion (lobar ICH, lobar CMB, cSS focus, or cSAH), OR (b) 1 white-matter feature alone (severe CSO-PVS >20 in one hemisphere, or multispot WMH pattern with >10 small subcortical FLAIR dots bilaterally) |
No deep hemorrhagic lesions and no other apparent cause of hemorrhagic lesions* |
Abbreviations: CAA, cerebral amyloid angiopathy; CMB, cerebral microbleed; cSAH, convexity subarachnoid hemorrhage; CSO-PVS, perivascular spaces in the centrum semiovale; cSS, cortical superficial siderosis; FLAIR, fluid-attenuated inversion recovery; ICH, intracerebral hemorrhage; SWI, susceptibility-weighted imaging; TFNE, transient focal neurological episode; WMH, white-matter hyperintensity.
Imaging
Gradient-echo MRI is one of the most important techniques used for diagnosis. Gradient-echo MRI demonstrates regions of low-signal blooming artifact caused by iron deposition left by old hemorrhages. See Image. Disseminated Cerebral Amyloid Angiopathies. Another beneficial technique is a susceptibility-weighted MRI (more sensitive than gradient-echo). Using these techniques, multiple areas of hemorrhage can be identified to help support the diagnosis.[6]
Pathology
Brain biopsies are rarely used for diagnosing CAA, except when CAA-related inflammation is suspected. If a sample is obtained, it should be examined with a β-amyloid immunostain or a Congo red stain. Using these techniques, vascular deposition of amyloid β-peptide can be identified, supporting the diagnosis of CAA.[7]
Treatment / Management
Management is often based on presenting symptoms. Acute management of patients with an intracerebral hemorrhage (ICH) is similar to the management of other spontaneous ICHs. Attention to blood pressure and intracranial pressure improves outcomes. When surgical intervention is performed, the mortality risk is comparable to other types of ICH. The factors that are associated with a worse prognosis are intraventricular hemorrhage and age older than 75 years.
ICH associated with CAA is frequently recurrent. Because of the high recurrence rate, clinicians typically avoid antiplatelet agents and anticoagulants when there is no strong indication for anticoagulation. Decisions about antithrombotic and anticoagulation therapy are individualized, and left atrial appendage occlusion is increasingly considered when cardioembolic risk is high and anticoagulation is prohibitively unsafe. While hemorrhagic complications may occur less frequently with novel oral anticoagulants than with warfarin, this finding may not apply to patients at higher risk of ICH, as these patients were excluded from all trials comparing novel oral anticoagulants to warfarin.[8][9](A1)
Blood pressure control reduces mortality, even though CAA is not driven by hypertension.[10] The Perindopril Protection Against Recurrent Stroke (PROGRESS) trial demonstrated a 77% reduction in CAA-related ICH with blood pressure control. This observation supports the finding that lower blood pressure is associated with a reduced risk of ICH recurrence. Lastly, limited evidence suggests that immunosuppression may benefit inflammatory forms of CAA. Results from small trials have shown that pulsed cyclophosphamide or glucocorticoids can lead to sustained clinical improvement. Other immunosuppressive medications have also demonstrated efficacy, including azathioprine, mycophenolate mofetil, and methotrexate.[11](A1)
Management of Transient Focal Neurological Episodes
Evidence to guide the treatment of TFNEs in CAA is limited. Initial management includes obtaining imaging to evaluate for new intracranial hemorrhage or convexity subarachnoid hemorrhage and controlling blood pressure appropriately. For recurrent episodes with an underlying mechanism of cortical spreading depolarizations (potentially arising from areas of cortical superficial siderosis and associated with acute convexity subarachnoid hemorrhage), a trial of antiseizure medications (such as valproic acid, lamotrigine, or topiramate) may be considered. However, evidence supporting the use of these medications in this setting is mainly anecdotal. Importantly, TFNEs are often mistaken for transient ischemic attacks. In patients presenting with focal transient deficits, gradient-echo or susceptibility-weighted MRI should be considered to look for cerebral microbleeds and cortical superficial siderosis. When TFNEs due to CAA are the more likely diagnosis, antithrombotic therapy should not be initiated or intensified, as it may increase the risk of intracranial hemorrhage.[12]
Differential Diagnosis
Nontraumatic ICH includes the following potential causes:
- Hemorrhagic tumor
- Hemorrhagic transformation of an ischemic stroke
- Lobar extension of a hypertensive putaminal hemorrhage
- Arteriovenous malformation
Other imaging differential diagnosis includes:
- Hypertensive microangiopathy
- Hemorrhagic metastases
- Multiple cavernoma syndrome
- Diffuse axonal injury
- Radiation-induced vasculopathy
- Neurocysticercosis
Prognosis
The prognosis of cerebral amyloid angiopathy is variable but is primarily determined by the location and size of the ICH. Unfavorable outcomes are associated with larger hematoma size and age (≥ 75 years). Favorable ICH outcomes are associated with sparing of the ventricles and a superficial location. Mortality ranges from 10% to 30%, with the best prognosis in patients with a higher level of consciousness and smaller hematomas (< 50 mL). CAA has a high hemorrhage recurrence risk when compared to a hypertensive hemorrhage. Study findings have revealed recurrence rates of approximately 21%.[13]
CAA is commonly associated with both transient and cognitive neurologic impairment. Transient symptoms are characterized by recurrent spells of numbness, paresthesia, and weakness, and the pathogenesis is not completely understood. Cognitive impairments include diminished cognitive speed and episodic memory loss. Interestingly, Alzheimer disease and CAA frequently coexist. Finally, CAA has also been associated with vascular dementia.[14]
Complications
Besides ICH, cerebral amyloid angiopathy has several other complications:
- Transient neurologic symptoms
- Cerebral microbleeds
- Cortical superficial siderosis
- CAA-related inflammation
- Cognitive impairment
CAA–Related Inflammation
A subset of patients with CAA develops spontaneous inflammatory episodes. This CAA-related inflammation (CAA-RI) most often presents with headache, seizures, focal neurological deficits, and a subacute or acute decline in cognition. Typical MRI findings include: (1) asymmetric white matter hyperintensities extending into the subcortical white matter, (2) hemorrhagic lesions such as cerebral microbleeds and cortical superficial siderosis, and (3) leptomeningeal enhancement following contrast administration. Cerebrospinal fluid examination is usually consistent with an inflammatory process, often showing a lymphocytic pleocytosis and elevated protein levels, although these abnormalities are not universally present. Notably, anti–amyloid-β antibodies can be detected in the cerebrospinal fluid during acute CAA-RI, supporting the concept of a spontaneous immune response to vascular amyloid-β. The combined clinical and radiographic findings closely resemble amyloid-related imaging abnormalities seen in patients receiving antiamyloid immunotherapies (see Table 2).[15]
Table 2. Diagnostic Criteria for CAA–Related Inflammation (CAA-RI)
|
Diagnosis |
Criteria |
|
Probable CAA-RI |
Patient age ≥ 40 years |
|
|
Presence of ≥ 1 of the following clinical features: headache, decrease in consciousness, behavioral change, or focal neurological deficits and seizures; the presentation is not directly attributable to an acute intracerebral hemorrhage |
|
|
MRI shows uni- or multifocal white matter hyperintensities extending to the immediate subcortical white matter; the asymmetry is not due to past ICH |
|
|
Presence of ≥ 1 of the following corticosubcortical hemorrhagic lesions: cerebral macrobleed, cerebral microbleed, or cortical superficial siderosis |
|
|
Exclusion of differential diagnoses (eg, neoplasm, infections) |
|
Possible CAA-RI |
Patient age ≥ 40 years |
|
|
Presence of ≥ 1 of the following clinical features: headache, decrease in consciousness, behavioral change, or focal neurological deficits and seizures; the presentation is not directly attributable to an acute intracerebral hemorrhage |
|
|
MRI shows white matter hyperintensities that extend to the immediate subcortical white matter |
|
|
Presence of ≥ 1 of the following corticosubcortical hemorrhagic lesions: cerebral macrobleed, cerebral microbleed, or cortical superficial siderosis |
|
|
Exclusion of differential diagnoses (eg, neoplasm, infections) |
Because CAA-RI is an autoimmune and inflammatory process, it often responds to immunosuppressive treatment. Most reports recommend starting with corticosteroid therapy, including high-dose methylprednisolone or dexamethasone, followed by a slow oral taper to reduce the risk of relapse and allow for sustained clinical stabilization. In more severe or recurrent cases, many authors recommend escalating to higher-potency immunosuppressants, most frequently combining corticosteroids with azathioprine or cyclophosphamide. Individual case reports and small series have also described the use of mycophenolate mofetil, methotrexate, rituximab, or intravenous immunoglobulin. Despite this range of therapeutic options, there are no established, objective criteria to guide the introduction of more aggressive immunosuppressive regimens.[16]
Deterrence and Patient Education
CAA is a cerebrovascular disorder characterized by the accumulation of amyloid β-protein within the leptomeninges and small- to medium-sized cerebral blood vessels.[1] Amyloid deposition can lead to fragile vessels that may cause lobar intracerebral hemorrhage (ICH). CAA may also present with cognitive impairments, incidental microbleeds, hemosiderosis, inflammatory leukoencephalopathy, Alzheimer disease, or transient neurological symptoms.[2] CAA can occur in specific familial syndromes or sporadically. Diagnosis is based on a combination of clinical, pathologic, and radiographic findings. Currently, no disease-modifying treatments are available. Prognosis depends on the presenting features of CAA, with worse outcomes in patients with large hematomas and in older adults.
Pearls and Other Issues
Iatrogenic cerebral amyloid angiopathy (iCAA) is a recently recognized form of CAA that differs from the typical age-related variant by presenting much earlier in life, most often in the third to fifth decades. Affected patients commonly develop recurrent intracerebral hemorrhage, seizures, and cognitive impairment. iCAA results from prior exposure to cadaveric biological materials, usually lyophilized dural grafts used in neurosurgical procedures, and shares mechanisms with prion diseases, in which misfolded proteins seed Aβ deposition in the brain. Diagnosing iCAA relies heavily on a detailed medical history to identify potential sources of exogenous Aβ. Clinical manifestations often occur decades after the index operation, with a typical latency of 30 to 40 years. Clinicians, especially neurosurgeons and vascular neurologists, should consider iCAA in younger patients who present with spontaneous lobar or otherwise atypical ICH suggestive of CAA, particularly when there is a history of cranial surgery involving cadaveric tissue (eg, lyophilized dura) or other procedures that could have introduced human Aβ. Given the association between cadaveric dural grafts and iCAA, the use of synthetic alternatives is strongly recommended.[17]
Enhancing Healthcare Team Outcomes
Management of cerebral amyloid angiopathy should involve an interprofessional team, including primary care clinicians, neurosurgeons, radiologists, and pathologists. Nurses specializing in stroke care, physical therapists, and pharmacists should also assist with the case. The patient most often will present to the emergency department with stroke symptoms. Using clinical and radiographic findings, the probability of CAA can be assessed. Treatment of ICH is similar to that of other spontaneous ICH, possibly requiring neurosurgical intervention. Attention to blood pressure and intracranial pressure is essential. If there is residual impairment, rehabilitation is necessary to regain mobility and function. The pharmacist may manage pain and help ensure appropriate use of antiplatelet agents and anticoagulants. Long-term follow-up is essential to evaluate for recurrence and functional status. If CAA is familial, further workup can be pursued in an outpatient setting. Open communication among interprofessional team members is essential for improving outcomes.
Media
(Click Image to Enlarge)
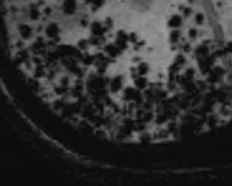
Disseminated Cerebral Amyloid Angiopathies. Gradient-echo MRI demonstrates several low-attenuation, blooming lesions, representing old iron deposits and hemorrhage.
Contributed by S Munakomi, MD
References
Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Annals of neurology. 2011 Dec:70(6):871-80. doi: 10.1002/ana.22516. Epub [PubMed PMID: 22190361]
Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. Journal of neurology, neurosurgery, and psychiatry. 2012 Feb:83(2):124-37. doi: 10.1136/jnnp-2011-301308. Epub 2011 Nov 5 [PubMed PMID: 22056963]
Level 3 (low-level) evidenceMiller-Thomas MM, Sipe AL, Benzinger TL, McConathy J, Connolly S, Schwetye KE. Multimodality Review of Amyloid-related Diseases of the Central Nervous System. Radiographics : a review publication of the Radiological Society of North America, Inc. 2016 Jul-Aug:36(4):1147-63. doi: 10.1148/rg.2016150172. Epub [PubMed PMID: 27399239]
Hu H, Wan S, Hu Y, Wang Q, Li H, Zhang N. Deciphering the role of APOE in cerebral amyloid angiopathy: from genetic insights to therapeutic horizons. Annals of medicine. 2025 Dec:57(1):2445194. doi: 10.1080/07853890.2024.2445194. Epub 2025 Jan 2 [PubMed PMID: 39745195]
Smith EE, Charidimou A, Ayata C, Werring DJ, Greenberg SM. Cerebral Amyloid Angiopathy-Related Transient Focal Neurologic Episodes. Neurology. 2021 Aug 3:97(5):231-238. doi: 10.1212/WNL.0000000000012234. Epub 2021 May 20 [PubMed PMID: 34016709]
Cheng AL, Batool S, McCreary CR, Lauzon ML, Frayne R, Goyal M, Smith EE. Susceptibility-weighted imaging is more reliable than T2*-weighted gradient-recalled echo MRI for detecting microbleeds. Stroke. 2013 Oct:44(10):2782-6. doi: 10.1161/STROKEAHA.113.002267. Epub 2013 Aug 6 [PubMed PMID: 23920014]
Level 2 (mid-level) evidenceChung KK, Anderson NE, Hutchinson D, Synek B, Barber PA. Cerebral amyloid angiopathy related inflammation: three case reports and a review. Journal of neurology, neurosurgery, and psychiatry. 2011 Jan:82(1):20-6. doi: 10.1136/jnnp.2009.204180. Epub 2010 Oct 9 [PubMed PMID: 20935328]
Level 3 (low-level) evidencePatel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, Breithardt G, Halperin JL, Hankey GJ, Piccini JP, Becker RC, Nessel CC, Paolini JF, Berkowitz SD, Fox KA, Califf RM, ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. The New England journal of medicine. 2011 Sep 8:365(10):883-91. doi: 10.1056/NEJMoa1009638. Epub 2011 Aug 10 [PubMed PMID: 21830957]
Level 1 (high-level) evidenceGranger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, Al-Khalidi HR, Ansell J, Atar D, Avezum A, Bahit MC, Diaz R, Easton JD, Ezekowitz JA, Flaker G, Garcia D, Geraldes M, Gersh BJ, Golitsyn S, Goto S, Hermosillo AG, Hohnloser SH, Horowitz J, Mohan P, Jansky P, Lewis BS, Lopez-Sendon JL, Pais P, Parkhomenko A, Verheugt FW, Zhu J, Wallentin L, ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. The New England journal of medicine. 2011 Sep 15:365(11):981-92. doi: 10.1056/NEJMoa1107039. Epub 2011 Aug 27 [PubMed PMID: 21870978]
Level 1 (high-level) evidenceArima H, Tzourio C, Anderson C, Woodward M, Bousser MG, MacMahon S, Neal B, Chalmers J, PROGRESS Collaborative Group. Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: the PROGRESS trial. Stroke. 2010 Feb:41(2):394-6. doi: 10.1161/STROKEAHA.109.563932. Epub 2009 Dec 31 [PubMed PMID: 20044530]
Level 1 (high-level) evidenceKinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, Greenberg SM. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007 Apr 24:68(17):1411-6 [PubMed PMID: 17452586]
Level 2 (mid-level) evidenceKozberg MG, Perosa V, Gurol ME, van Veluw SJ. A practical approach to the management of cerebral amyloid angiopathy. International journal of stroke : official journal of the International Stroke Society. 2021 Jun:16(4):356-369. doi: 10.1177/1747493020974464. Epub 2020 Nov 29 [PubMed PMID: 33252026]
Rosand J, Muzikansky A, Kumar A, Wisco JJ, Smith EE, Betensky RA, Greenberg SM. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Annals of neurology. 2005 Sep:58(3):459-62 [PubMed PMID: 16130107]
Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Annals of neurology. 2011 Feb:69(2):320-7. doi: 10.1002/ana.22112. Epub 2010 Nov 8 [PubMed PMID: 21387377]
Auriel E, Charidimou A, Gurol ME, Ni J, Van Etten ES, Martinez-Ramirez S, Boulouis G, Piazza F, DiFrancesco JC, Frosch MP, Pontes-Neto OV, Shoamanesh A, Reijmer Y, Vashkevich A, Ayres AM, Schwab KM, Viswanathan A, Greenberg SM. Validation of Clinicoradiological Criteria for the Diagnosis of Cerebral Amyloid Angiopathy-Related Inflammation. JAMA neurology. 2016 Feb:73(2):197-202. doi: 10.1001/jamaneurol.2015.4078. Epub [PubMed PMID: 26720093]
Level 1 (high-level) evidenceSeifert RM, Klingebiel R, Schäbitz WR. Diagnosis, pathomechanisms and therapy of cerebral amyloid angiopathy-related inflammation (CAA-ri). Neurological research and practice. 2025 Apr 26:7(1):26. doi: 10.1186/s42466-025-00382-3. Epub 2025 Apr 26 [PubMed PMID: 40281535]
Frol S, Splavski B. To reveal and acknowledge an iatrogenic cerebral amyloid angiopathy: a vascular neurologist and neurosurgeon collaborative perspective. Frontiers in neurology. 2025:16():1527417. doi: 10.3389/fneur.2025.1527417. Epub 2025 Jun 13 [PubMed PMID: 40584521]
Level 3 (low-level) evidence