Introduction
Bardet-Biedl syndrome (BBS; OMIM 209900) represents a rare autosomal recessive multisystem ciliopathy caused by pathogenic variants in genes encoding proteins essential for the structure and function of primary nonmotile cilia, situating BBS within the broader class of ciliopathies. Georges Bardet first described the condition in 1920, followed independently by Arthur Biedl in 1922, with subsequent unification of their observations under the current eponym.[1]
Clinically, BBS presents with 6 cardinal features, including rod-cone retinal dystrophy, early-onset obesity with hyperphagia, postaxial polydactyly, renal structural and functional anomalies, hypogonadism with genital anomalies, and neurodevelopmental and cognitive impairment. The phenotype demonstrates marked inter- and intrafamilial variability, with manifestations that evolve across the lifespan, from the prenatal period through adulthood.[2]
Diagnostic criteria for BBS have undergone major refinement in recent years. A 2024 collaborative consensus statement from the European Reference Networks (Inter-ERN) revised the original Beales 1999 criteria to integrate clinical features with molecular genetic findings.[3] A 2026 publication by Pomeroy and colleagues introduced a further refined diagnostic algorithm, offering a simplified, globally applicable framework that integrates major and minor clinical criteria with molecular genetic data.[2] A significant therapeutic advancement in BBS management occurred with FDA approval of setmelanotide (Imcivree), the first targeted pharmacotherapy for the condition. Initial approval in June 2022 included patients 6 years and older, with subsequent expansion in December 2024 to include children as young as 2 years.[4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
BBS is inherited in an autosomal recessive pattern. At least 26 genes have been definitively associated with the condition, each encoding a protein that localizes primarily to the basal body, centrosome, or primary cilium.[2] The BBSome, an octameric coat-like complex composed of BBS1, BBS2, BBS4, BBS5, BBS7, BBS8 (TTC8), BBS9, and BBS18 (BBIP1), mediates selective intraflagellar transport of G-protein-coupled receptors within the ciliary membrane. These receptors include the melanocortin-4 receptor (MC4R) and leptin receptor, both of which are central to appetite regulation and energy homeostasis. A second group of BBS proteins, including BBS6 (MKKS), BBS10, and BBS12, functions as chaperonin-like proteins that facilitate BBSome assembly.[5]
Pathogenic variants in BBS1 account for approximately 23% to 43% of cases, depending on the cohort, and the p.M390R missense variant is the most prevalent BBS1 allele in individuals of Northern European ancestry. BBS10 is the second most commonly implicated gene, accounting for approximately 25% of positive findings in the Clinical Registry Investigating Bardet-Biedl Syndrome (CRIBBS).[2] An oligogenic or modifier inheritance model has been proposed in selected kindreds, in which secondary variants in additional BBS-associated or ciliary genes appear to influence penetrance and severity. However, classical biallelic recessive inheritance at a single locus remains the predominant mechanism.[6] Founder effects have been described in several genetically isolated populations, including Bedouin communities in the Middle East, the Faroe Islands, Newfoundland, and the island of La Réunion, contributing to regionally elevated prevalence.[3]
Epidemiology
BBS affects approximately 1 in 100,000 to 1 in 160,000 individuals in North American and European populations. Prevalence is substantially higher in genetically isolated communities, reaching approximately 1 in 13,500 to 1 in 17,500 among Bedouin populations of Kuwait and the wider Middle East, and approximately 1 in 17,500 in Newfoundland, Canada. An estimated 1,500 to 2,500 individuals are affected in the United States.[3]
BBS affects males and females in equal proportions, with no established sex predilection. Diagnosis most commonly occurs in childhood. A recent multicountry cohort study reported a median age at confirmed diagnosis of approximately 9 years, reflecting the progressive nature of the syndrome and the variable age at which individual features become clinically apparent. The global burden of BBS is likely underestimated, particularly in low- and middle-income countries, where underrecognition of clinical features and limited access to molecular genetic testing contribute to underdiagnosis.[2]
Pathophysiology
BBS is a primary nonmotile ciliopathy that arises from dysfunction of proteins required for ciliary biogenesis, intraflagellar transport, and receptor trafficking at the primary cilium.[5] Although primary cilia are present on nearly every cell type in the human body, the clinical manifestations of BBS reflect the tissues in which ciliary signaling is most physiologically essential, including the hypothalamus, kidney, retina, and developing limb. Disruption of BBSome function impairs ciliary trafficking of the leptin receptor to the surface of hypothalamic arcuate nucleus pro-opiomelanocortin (POMC) neurons.
The resulting attenuation of leptin signaling leads to deficient release of alpha-melanocyte-stimulating hormone (α-MSH), the endogenous agonist of the melanocortin-4 receptor (MC4R) in the paraventricular nucleus. Impaired MC4R activation in turn produces severe, early-onset hyperphagia and obesity that is pathobiologically distinct from common polygenic obesity and is largely refractory to conventional dietary and lifestyle management. This mechanism provides the biological rationale for setmelanotide, an MC4R agonist that bypasses the upstream signaling defect.[2]
In the kidney, ciliary dysfunction within tubular epithelial cells disrupts tubulogenesis and mechanosensory flow detection, contributing to structural anomalies including cortical and medullary cysts, calyceal malformations, and parenchymal dysplasia. These changes can progress over time to chronic kidney disease and, in a subset of patients, to kidney failure requiring renal replacement therapy. In the retina, failure of ciliary transport across the photoreceptor connecting cilium leads to mislocalization of opsins and progressive rod-cone dystrophy, beginning with rod-mediated night blindness in childhood and progressing to cone involvement, peripheral and central visual field loss, and, in most patients, legal blindness by the second or third decade of life.[3]
History and Physical
Per the 2026 updated consensus diagnostic criteria proposed by Pomeroy and colleagues, clinical manifestations are categorized into 6 major criteria and 8 minor criteria (see Image. Diagnostic Criteria for Bardet-Biedl Syndrome). A clinical diagnosis of BBS is supported by the presence of either 4 major criteria or 3 major criteria plus 2 minor criteria. Importantly, the absence of any single criterion does not exclude the diagnosis, because not all manifestations are present in every patient, and the age of onset varies considerably across organ systems.[2]
Major Criterion 1: Retinal Dystrophy
Retinal dystrophy is the most consistent feature across genotypes and is present in approximately 76% to 97% of individuals, with prevalence increasing with age. BBS-related retinal disease manifests predominantly as a rod-cone dystrophy. Early rod-mediated symptoms include nyctalopia, which is typically apparent by 7 to 8 years of age, and progressive peripheral visual field constriction. These findings later progress to cone dysfunction with reduced central visual acuity and color discrimination. Additional early features, eg, scotopic adaptation defects, blurred or distorted vision, and nystagmus, should prompt formal ophthalmologic evaluation, including electroretinography. Legal blindness commonly develops between the second and fourth decades of life.[2][1]
Major Criterion 2: Obesity or Overweight
Obesity is present in approximately 87% to 89% of individuals with BBS and typically begins within the first few years of life. Obesity at any age qualifies as a major criterion, defined as a body mass index (BMI) of 30 kg/m² or greater in adults, and a BMI at or above the 95th percentile for age and sex in pediatric patients 2 years and older. In children younger than 2 years, weight-for-length at or above the 85th percentile is considered a major criterion, enabling earlier recognition. Hyperphagia, characterized by abnormal food-seeking behaviors, inability to achieve satiety, and persistent preoccupation with food, is present in nearly all affected individuals. However, hyperphagia is not itself counted as a separate diagnostic criterion because no standardized, validated assessment tool is currently in routine clinical use.[2][7]
Major Criterion 3: Congenital Anomalies of the Kidney and Urinary Tract or Chronic Kidney Disease
Renal involvement is present in approximately 26% to 81% of patients, depending on the specific anomaly assessed, and chronic kidney disease (CKD) is reported in 31% to 39% of affected individuals. Congenital anomalies of the kidney and urinary tract (CAKUT) in BBS encompass renal hypoplasia or agenesis, renal cysts, fetal lobulation, caliectasis, calyceal clubbing, loss of renal corticomedullary differentiation, and ectopic kidney. When both CAKUT and CKD are present in the same patient, they are counted only once toward the diagnostic threshold. Advanced CKD progressing to kidney failure requiring dialysis or transplantation occurs in approximately 6% to 7% of patients and represents a major contributor to BBS-associated morbidity and mortality.[2][8]
Major Criterion 4: Hypogonadism or Genital Anomalies
Hypogonadism or genital anomalies are present in approximately 34% to 68% of patients and are counted once when both are present. In males, findings include micropenis (approximately 70%), cryptorchidism (approximately 26% to 28%), and delayed puberty. In females, findings include vaginal atresia (approximately 14% to 22%), hydrocolpos or hydrometrocolpos (approximately 8% to 35%), hypoplastic or duplex uterus, and persistent urogenital sinus. These features may be the earliest recognized manifestations in the neonatal period and warrant prompt evaluation by pediatric urology and endocrinology.[2]
Major Criterion 5: Neurodevelopmental and Cognitive Manifestations
Neurodevelopmental and cognitive manifestations are present in approximately 67% to 72% of individuals and are formally recognized as a major diagnostic criterion in the 2026 framework. This represents a meaningful update from prior criteria and reflects both the high prevalence and the diagnostic value of these features. They are counted only once regardless of the number of associated manifestations identified in a given patient.
Specific findings include delays in expressive language and fine motor development, intellectual disability (Full Scale IQ consistent with intellectual disability in 20% to 58% of patients), features of autism spectrum disorder (approximately 35.6% screen positive, with 17% receiving a formal diagnosis), and imbalance or ataxia. The absence of neurodevelopmental dysfunction does not preclude a BBS diagnosis.[2]
Major Criterion 6: Postaxial Polydactyly
Postaxial polydactyly is present in approximately 75% to 79% of individuals with BBS. Postaxial polydactyly may be symmetrical or asymmetrical and may affect 1 or more extremities. This finding is most often identified prenatally on routine obstetric ultrasonography or at birth, and is frequently surgically removed in infancy. In older patients, careful examination for surgical scar remnants along the ulnar or fibular borders, along with a detailed birth history, is essential for documenting this feature.[2]
Minor Criteria
Eight minor criteria are recognized in the 2026 framework: urine concentration defects at any age, or chronic urinary retention in adults (not counted separately when CAKUT or CKD is already counted); brachydactyly or syndactyly (approximately 56%); hearing loss, predominantly conductive (approximately 11%); laterality defects (approximately 1%); anosmia or hyposmia (approximately 41%); dental anomalies; craniofacial dysmorphisms; and metabolic dysfunction-associated steatotic liver disease (MASLD, not counted separately when obesity is already counted).[2]
Age-Dependent Presentation
The clinical phenotype of BBS evolves substantially across the lifespan. In the prenatal period, sonographic findings may include postaxial polydactyly and renal hyperechogenicity. In the neonatal period, polydactyly and genitourinary anomalies are frequently the presenting features. Obesity and hyperphagia typically manifest by 1 to 2 years of age. Retinal dystrophy becomes functionally evident in mid-childhood, often after the diagnosis of polydactyly and obesity has prompted closer surveillance. Adolescents and adults face compounding morbidity from progressive visual impairment, advancing renal disease, hypogonadism, and metabolic complications, including type 2 diabetes and dyslipidemia.[8]
| Pause and Reflect |
A 6-year-old boy is referred for evaluation of difficulty seeing at night. His parents report that he was born with extra fingers on both hands, which were surgically removed in infancy. He has been significantly overweight since approximately 18 months of age, despite careful dietary management at home, and he frequently takes food from the kitchen without permission. His body mass index (BMI) is at the 99th percentile for age and sex. Fundoscopic examination reveals bilateral bone-spicule pigmentation and attenuated retinal vessels.
|
Evaluation
Diagnostic Algorithm
Per the 2026 consensus algorithm, all patients with clinical suspicion of BBS, defined as the presence of 1 or more characteristic clinical manifestations or a positive family history, should undergo both clinical evaluation and molecular genetic testing where available. A clinical diagnosis is established when a patient meets 4 major criteria or 3 major criteria plus 2 minor criteria. When clinical criteria are met, genetic confirmation is not required to establish the diagnosis. However, molecular testing remains valuable for management decisions, family planning, and access to targeted therapies (see Image. Bardet-Biedl Syndrome Diagnostic Algorithm).[2]
Genetic Testing and Interpretation
Molecular genetic testing is strongly recommended for all patients, including those who already meet clinical criteria, because the results guide treatment eligibility, inform genetic counseling, and support clinical trial enrollment. Next-generation sequencing panels that include all confirmed BBS-associated genes are the preferred testing modality. Whole-exome or whole-genome sequencing may be considered when panel testing is inconclusive. A positive genetic result is defined as biallelic pathogenic or likely pathogenic variants in a recognized BBS gene, and when combined with at least 1 major clinical criterion, establishes the diagnosis.
An inconclusive result occurs when testing identifies biallelic pathogenic or likely pathogenic variants together with a variant of uncertain significance (VUS), biallelic VUS, or a single heterozygous pathogenic or likely pathogenic variant. A negative result is defined as no variants of likely relevance identified. An inconclusive or negative genetic result does not exclude BBS, because approximately 10% to 20% of patients with a confirmed clinical diagnosis do not have an identified pathogenic genotype on currently available testing. Variant classifications should be reviewed periodically, since reclassification of variants over time, particularly the reclassification of a VUS to pathogenic or likely pathogenic, may convert an inconclusive result into a diagnostic one.[2][6]
Ophthalmologic Evaluation
Formal ophthalmologic assessment at diagnosis should include electroretinography, optical coherence tomography, fundus autofluorescence imaging, and visual field testing. Electroretinography can demonstrate reduced or absent rod and cone responses before patients report subjective visual symptoms and is therefore central to early diagnosis and prognostic counseling. Annual ophthalmologic surveillance is recommended, with the frequency and specific testing modalities individualized based on age and disease stage. Low-vision rehabilitation and orientation-and-mobility services should be introduced early in patients with progressive visual loss.[1]
Renal Evaluation
Renal ultrasonography at diagnosis is used to document structural anomalies, including cysts, dysplasia, and collecting-system abnormalities. Baseline and annual laboratory evaluation should include serum creatinine, cystatin C, estimated glomerular filtration rate (eGFR), urinalysis with microscopy, and urine protein-to-creatinine ratio. Blood pressure monitoring is essential, as hypertension is common and contributes to CKD progression. Early referral to pediatric or adult nephrology is recommended for patients with structural anomalies, reduced eGFR, proteinuria, or hypertension.[8]
Metabolic, Endocrine, Cardiac, and Neurodevelopmental Evaluation
Fasting glucose, hemoglobin A1c, fasting lipid panel, liver function tests, and thyroid function should be assessed at diagnosis and monitored regularly to identify and manage metabolic complications, including type 2 diabetes, dyslipidemia, and metabolic dysfunction-associated steatotic liver disease. Gonadal function should be evaluated with luteinizing hormone (LH), follicle-stimulating hormone (FSH), and morning total testosterone in males, and with pelvic ultrasonography in females. Clinical findings and pubertal stage should guide any additional evaluation of female hormones.
Baseline echocardiography is recommended in all patients at diagnosis to screen for congenital structural heart disease, with subsequent imaging guided by symptoms and initial findings. Formal neuropsychological and developmental assessment should be performed in all pediatric patients, including standardized screening for autism spectrum disorder. Repeat assessments at key developmental and educational transitions are recommended to support individualized education planning and to identify emerging behavioral or psychiatric comorbidities.[5]
Treatment / Management
Setmelanotide (Imcivree)
Setmelanotide, a melanocortin-4 receptor (MC4R) agonist, is the first and currently only FDA-approved pharmacotherapy for obesity management in BBS. Setmelanotide is approved for patients 2 years and older with obesity attributable to BBS, following the December 2024 age expansion from the prior indication of 6 years and older, which was originally granted in June 2022. In the pivotal phase 3 trial, setmelanotide produced a mean BMI reduction of approximately 7.9% at 52 weeks in the open-label period, and a 4.6% reduction compared with a 0.1% reduction for placebo during the 14-week double-blind stage, along with statistically significant reductions in hunger scores.[4](A1)
Mechanistically, setmelanotide directly targets the underlying MC4R pathway impairment. Restoration of agonist activity at the hypothalamic MC4R bypasses the upstream leptin-melanocortin signaling defect that drives hyperphagia in BBS. Common adverse effects include skin hyperpigmentation (attributable to off-target MC1R agonism), injection site reactions, nausea, headache, and diarrhea. The prescribing information includes warnings for serious psychiatric adverse events, including depression and suicidal ideation, and patients should be monitored accordingly during treatment (see Image. Setmelanotide Mechanism of Action in Bardet-Biedl Syndrome).[9](B3)
Obesity and Metabolic Management
Comprehensive obesity management in BBS requires an interprofessional approach. Dietary counseling and behavioral strategies should be tailored to BBS-associated hyperphagia, with attention to structured mealtimes, environmental modifications to limit unsupervised access to food, and family-based behavioral interventions. Physical activity programming should be adapted for visual impairment, balance difficulties, and orthopedic considerations. Bariatric surgery has been reported in small case series with variable outcomes, and given the underlying neurobiological drive for hyperphagia, durable weight loss after surgery alone is often limited. An individualized risk-benefit assessment, with input from an interprofessional obesity team, is recommended.[5]
Renal Management
Renal disease in BBS is comanaged with nephrology. Hypertension should be identified and treated promptly, with renin-angiotensin-aldosterone system (RAAS) inhibition using an angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB), with the latter preferred in the setting of proteinuria. Patients should be monitored for progression of CKD with periodic eGFR and urinary albumin assessment, and timely planning for kidney replacement therapy should be initiated when appropriate. Renal transplantation outcomes in BBS are reported to be comparable to those in other genetic nephropathies, although recipients require careful posttransplant management of obesity and metabolic comorbidities.[8]
Ophthalmologic Management
Currently, no disease-modifying therapy has been approved for BBS-associated retinal dystrophy. Supportive care, including low-vision aids, orientation and mobility training, educational accommodations, and early connection with vision rehabilitation services, is central to preserving function and quality of life. Investigational gene therapy is in early clinical development. AXV-101 (Axovia Therapeutics), an AAV9-based gene therapy delivering a codon-optimized BBS1 transgene, has received FDA Orphan Drug and Rare Pediatric Disease designations. A first-in-human, open-label, dose-escalation Phase 1/2 trial (AXIS; NCT07269665) was registered in early 2026 to evaluate safety, tolerability, and pharmacodynamics in patients with biallelic BBS1 pathogenic variants and retinal degeneration.[1](B3)
Endocrine and Reproductive Management
Confirmed hypogonadotropic hypogonadism is managed with sex hormone replacement therapy, individualized to the patient's age, pubertal status, and reproductive goals. Type 2 diabetes mellitus is managed per current guidelines, with consideration of agents that offer cardiorenal benefits, eg, sodium-glucose cotransporter-2 (SGLT2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists, when appropriate for the individual patient. Dyslipidemia, metabolic dysfunction-associated steatotic liver disease, and thyroid dysfunction should be screened for and managed in parallel.[3] (B3)
| Pause and Reflect |
The parents of a 3-year-old girl with genetically confirmed BBS, due to a homozygous pathogenic variant in BBS10, present to the clinic in distress because of their daughter's uncontrollable food-seeking behavior. She wakes at night to search for food, has gained 8 kg over the past year, and has a BMI z-score of +3.5. The family has implemented strict dietary restrictions at home but reports constant conflict around food, escalating frustration, and significant family stress. Both parents express guilt and exhaustion, and the child's older sibling has begun to express resentment about the family's focus on food.
|
Differential Diagnosis
Several conditions share overlapping features with BBS and must be distinguished through careful clinical evaluation and molecular genetic testing. BBS also has significant genetic and clinical overlap with other ciliopathies, and the possibility of allelic disorders should be considered when variants in BBS-associated genes are identified.[2]
Alström syndrome is an autosomal recessive ciliopathy caused by biallelic pathogenic variants in ALMS1. This syndrome presents with rod-cone retinal dystrophy, childhood obesity, sensorineural hearing loss, dilated cardiomyopathy, and severe insulin resistance. Alström syndrome is distinguished from BBS by the presence of cardiomyopathy and hearing loss, the absence of postaxial polydactyly, and typically preserved cognitive function.
Laurence-Moon syndrome is now considered a distinct entity from BBS, characterized by progressive spastic paraplegia rather than polydactyly, and has been associated with biallelic variants in PNPLA6. The historical eponym "Laurence-Moon-Bardet-Biedl syndrome" reflects earlier conflation of the 2 conditions and is no longer recommended. Prader-Willi syndrome presents with neonatal hypotonia and feeding difficulties, followed in early childhood by hyperphagia, obesity, hypogonadism, and intellectual disability. Prader-Willi syndrome is caused by loss of paternally expressed genes in the 15q11-q13 region and is confirmed by DNA methylation analysis. Retinal dystrophy and polydactyly are not features that distinguish it from BBS.
McKusick-Kaufman syndrome (MKKS) is caused by biallelic variants in the MKKS gene, also designated BBS6, and is therefore an allelic disorder of BBS. MKKS is characterized by postaxial polydactyly, hydrometrocolpos, congenital heart defects, and genitourinary malformations, while retinal dystrophy, obesity, and neurodevelopmental impairment are typically absent. This distinction is clinically important when biallelic MKKS/BBS6 variants are identified, because the same genotype can produce either MKKS or BBS, depending on the variant and modifying factors. Longitudinal follow-up may be required before the phenotype is clear, particularly in infancy.[2]
Joubert syndrome is distinguished by cerebellar vermis hypoplasia producing the pathognomonic "molar tooth sign" on brain MRI, along with hypotonia, oculomotor apraxia, and abnormal breathing patterns in infancy. Renal and retinal features overlap in BBS, and several ciliary genes are shared between the 2 conditions, reinforcing the value of molecular genetic testing in ambiguous presentations. Cohen syndrome is caused by biallelic variants in VPS13B and features intellectual disability, characteristic facial dysmorphism, truncal obesity, microcephaly, and intermittent neutropenia. The retinal dystrophy seen in Cohen syndrome differs in pattern from the rod-cone dystrophy of BBS, and postaxial polydactyly is not a feature.[1]
Prognosis
The prognosis of BBS is determined largely by the severity and trajectory of renal disease, which remains the leading cause of premature mortality. Chronic kidney disease progresses to kidney failure in a meaningful proportion of patients, with approximately 6% to 7% ultimately requiring kidney replacement therapy in the form of dialysis or transplantation.[2] Progressive retinal dystrophy follows a near-universal course, and most patients reach legal blindness between the second and fourth decades of life.
Cardiovascular morbidity related to obesity, hypertension, dyslipidemia, and metabolic syndrome contributes meaningfully to long-term outcomes and is increasingly recognized as a target for early intervention. Neurodevelopmental trajectories are variable. Intellectual disability and behavioral comorbidities are common, but many individuals achieve meaningful functional independence with timely educational support, behavioral therapy, and family-centered care.
The availability of setmelanotide offers the potential to mitigate the metabolic and quality-of-life consequences of hyperphagia and early-onset obesity when initiated early in the disease course. However, long-term outcome data on renal, cardiovascular, and survival endpoints are not yet available, and clinical registries eg, CRIBBS and the European BBS networks, will be essential to define these outcomes over time.[1][8]
Complications
BBS is associated with multiorgan complications that accumulate across the lifespan. The most consequential and life-limiting of these is progressive CKD, which may advance to kidney failure and remains the primary cause of premature mortality. Vision loss progressing to legal blindness profoundly affects quality of life, educational attainment, vocational opportunities, and overall functional independence and warrants early integration of low-vision rehabilitation and assistive services.
Obesity-related complications are common and frequently develop in childhood or adolescence. These include type 2 diabetes, metabolic syndrome, dyslipidemia, obstructive sleep apnea, and metabolic dysfunction-associated steatotic liver disease (MASLD), which may progress silently to hepatic fibrosis in a subset of patients. Hypertension is common and accelerates both cardiovascular and renal disease progression.
Congenital heart defects, including atrial and ventricular septal defects, patent ductus arteriosus, and left ventricular hypertrophy, are recognized cardiac associations and underscore the rationale for baseline echocardiography at diagnosis. Reproductive complications include infertility, hypogonadotropic hypogonadism, and structural genitourinary anomalies that may require surgical management. Neuropsychiatric manifestations, including anxiety, depression, features of autism spectrum disorder, obsessive-compulsive traits, and emotional dysregulation, are increasingly recognized as core contributors to social functioning and quality of life, and should be addressed proactively as part of comprehensive care.[5][1]
Consultations
Comprehensive BBS management requires coordinated input from multiple subspecialties and allied health professionals working together as an interprofessional team. Care is most effective when a primary care clinician or designated medical home coordinator helps the patient and family navigate referrals, surveillance schedules, and transitions between pediatric and adult care.
Ophthalmology should be engaged from the time of diagnosis for baseline assessment, retinal surveillance, and timely referral to low-vision rehabilitation, orientation and mobility training, and assistive technology services as visual function declines. Nephrology is essential for the evaluation of structural and functional renal disease, the annual monitoring of kidney function and blood pressure, the management of hypertension and proteinuria, and the planning for kidney replacement therapy when needed. Pediatric or adult endocrinology manages obesity and hyperphagia, including initiation and monitoring of setmelanotide. Endocrinology also addresses type 2 diabetes, dyslipidemia, hypogonadotropic hypogonadism, and thyroid dysfunction. Bariatric or obesity medicine specialists may be involved when surgical or intensive medical weight management is being considered.
Medical genetics provides molecular diagnostic confirmation, variant interpretation, periodic reviews of variant reclassification, and lifelong genetic counseling for patients and family members, including reproductive counseling and prenatal options. Developmental pediatrics, neurodevelopmental specialists, or clinical neuropsychology address neurodevelopmental and neuropsychiatric comorbidities. This includes formal cognitive assessment, autism spectrum disorder evaluation, and management of anxiety, depression, and behavioral concerns. Child and adolescent psychiatry may be involved when pharmacologic treatment of psychiatric symptoms is needed.[3][10]
Registered dietitians with experience in hyperphagia and rare genetic obesity provide individualized nutritional counseling, anticipatory guidance for families, and coordination with behavioral interventions. Urology and gynecology address structural genitourinary anomalies, including surgical management of hydrometrocolpos, cryptorchidism, vaginal atresia, and related conditions. Reproductive endocrinology and fertility specialists are consulted when reproductive goals are being discussed. Cardiology is engaged for baseline echocardiography at diagnosis and for management of congenital heart disease, hypertension, and obesity-related cardiovascular complications.
Audiology evaluates and monitors conductive and sensorineural hearing loss, which is reported in a subset of patients with BBS and may be more common in those with chronic otitis media. Sleep medicine and pulmonology assess and manage obstructive sleep apnea, which is common in the setting of obesity and craniofacial anatomy. Dentistry, including pediatric dentistry and orthodontics, addresses the dental anomalies, malocclusion, and high palate frequently seen in BBS. Physical therapy, occupational therapy, and speech-language pathology support motor coordination, balance, activities of daily living, expressive and receptive language, and feeding skills. Social work and patient advocacy organizations, including the Bardet-Biedl Syndrome Foundation, provide families with peer support, educational resources, financial and insurance navigation, and connections to clinical registries such as CRIBBS and ongoing research opportunities.[3][10]
Deterrence and Patient Education
Genetic counseling is a cornerstone of BBS care and should address the 25% recurrence risk in future pregnancies, the availability of preimplantation genetic testing for couples planning future children, carrier testing for extended family members, and prenatal testing options when appropriate. Patient and family education must address the neurobiological basis of hyperphagia in BBS. The persistent food-seeking behavior and inability to feel full stem from dysfunction of the MC4R pathway in the hypothalamus, not from willful behavior, lack of discipline, or inadequate parenting. Framing hyperphagia accurately is essential to reduce stigma, prevent misattribution of symptoms, and support families in seeking appropriate medical and behavioral care.[2][7]
Practical strategies for managing hyperphagia in the home environment should be discussed proactively. These include securing food storage, establishing structured and predictable mealtimes, planning portion-controlled meals and snacks, and using family-based behavioral approaches that involve siblings and other household members rather than singling out the child with BBS. Anticipatory guidance should be provided well before behaviors become entrenched. Families should be educated about the progressive nature of retinal dystrophy and connected early with low-vision services, orientation and mobility training, educational accommodations, and assistive technology. Initiating these services before functional vision loss becomes advanced supports independence and continuity of learning and employment.
The period of diagnostic uncertainty that often precedes confirmation of BBS can impose substantial psychological and emotional costs on patients and families. Timely diagnosis allows families to access services, peer support, and clinical research opportunities from a position of clarity rather than continued searching. Patients and families should be connected with the Bardet-Biedl Syndrome Foundation, the National Organization for Rare Disorders (NORD), and relevant national and international rare disease networks. These organizations provide peer support, educational materials, family conferences, advocacy resources, and pathways to participate in registries such as CRIBBS and in emerging clinical trials.[2]
Enhancing Healthcare Team Outcomes
Optimal care for individuals with BBS requires a deliberately coordinated interprofessional team, reflecting the multisystem disease burden and the lifelong nature of management. The primary care clinician, whether a pediatrician, family physician, or internist, serves as the central hub of care coordination. This role includes ensuring that surveillance protocols are implemented, specialist recommendations are integrated, medications are reconciled, and transitions from pediatric to adult care are planned proactively and well in advance. The 2026 Pomeroy diagnostic algorithm reinforces the role of primary care in maintaining diagnostic vigilance across age groups. Clinical features in BBS may emerge incrementally over the years, and a negative or inconclusive genetic result does not exclude the diagnosis. Primary care clinicians are uniquely positioned to recognize evolving patterns and prompt re-evaluation when new features appear.[2][3]
Pediatric obesity medicine specialists and endocrinologists play a central role in evaluating eligibility for setmelanotide, initiating therapy, titrating the dose, and monitoring for both metabolic response and adverse effects. Pharmacists contribute through structured patient and caregiver education on subcutaneous injection technique, recognition of adverse effects (including skin hyperpigmentation, mood changes, and injection site reactions), medication storage and handling, and adherence support over time. Nursing staff play a critical role in monitoring hyperphagia severity, blood pressure, and growth, providing family-centered counseling, and engaging in empathetic, non-stigmatizing communication with patients and caregivers as they manage the psychosocial burden of a rare chronic disease.[9]
Ophthalmology and nephrology should maintain bidirectional communication with the primary care team regarding disease progression and cross-system management decisions, including the timing of low-vision referrals, blood pressure targets, and planning for kidney replacement therapy when indicated. Genetic counselors bridge the diagnostic and ongoing care phases, particularly as the BBS gene catalog continues to expand and variant classifications evolve. Developmental specialists, neuropsychologists, and child and adolescent psychiatry teams address the often underrecognized neurodevelopmental and neuropsychiatric burden, including features of autism spectrum disorder that may otherwise be misattributed to other causes or remain undiagnosed. At the health systems level, the healthcare team has a collective responsibility to advocate for equitable access to molecular genetic testing, to setmelanotide and other targeted therapies across payer landscapes, to early childhood screening for BBS features, and to inclusion of patients with BBS in clinical trials and registries. These advocacy efforts are particularly important for patients in low- and middle-income countries and for populations historically underrepresented in rare disease research.[2][9][10]
Media
(Click Image to Enlarge)
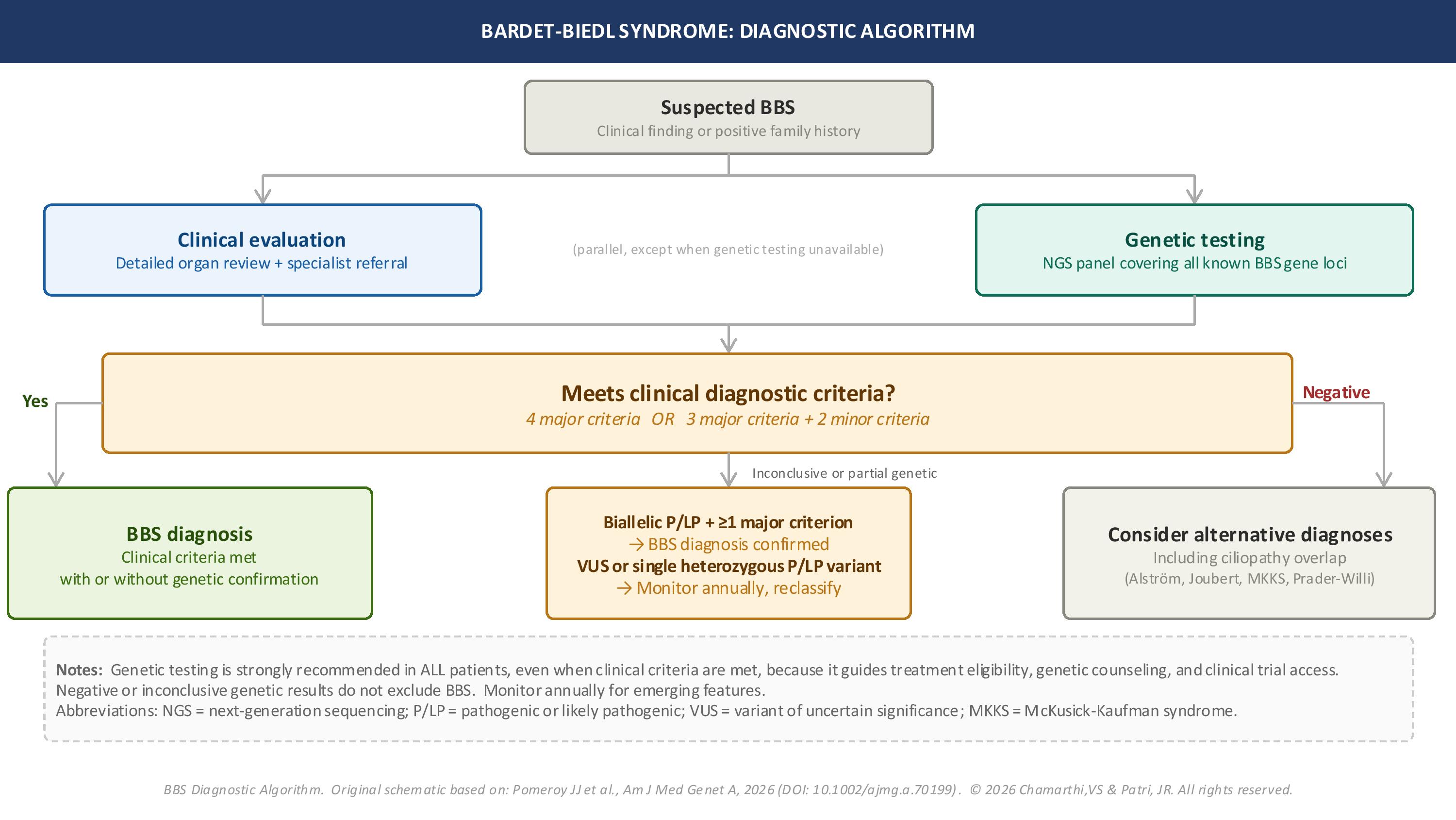
Diagnostic Algorithm for Bardet-Biedl Syndrome. The figure outlines clinical assessment, genetic testing, the application of major and minor diagnostic criteria, the interpretation of genetic results, and the consideration of alternative ciliopathy diagnoses. The algorithm incorporates the updated 2026 diagnostic framework and emphasizes parallel clinical and molecular evaluation.
Contributed by V Sushma Chamarthi, MD, and Jyothi R Patri, MD
(Click Image to Enlarge)
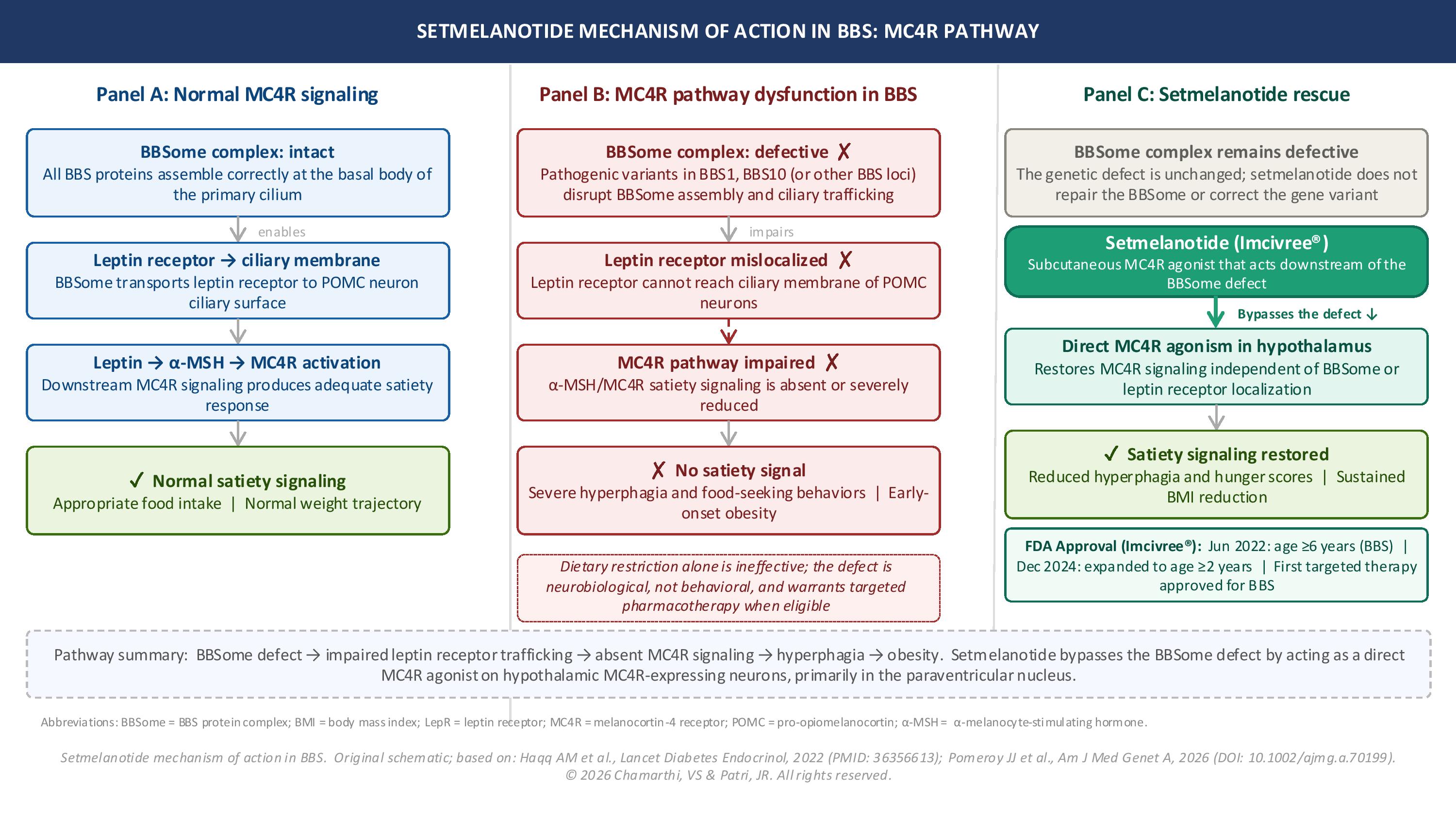
Setmelanotide Mechanism of Action in Bardet-Biedl Syndrome. Illustration of melanocortin-4 receptor (MC4R) signaling in Bardet-Biedl syndrome and the therapeutic mechanism of setmelanotide. The figure compares normal MC4R signaling, BBSome dysfunction associated with BBS, which impairs leptin receptor trafficking and satiety signaling, and the restoration of downstream MC4R activation by setmelanotide treatment.
Contributed by V Sushma Chamarthi, MD, and JR Patri, MD
(Click Image to Enlarge)
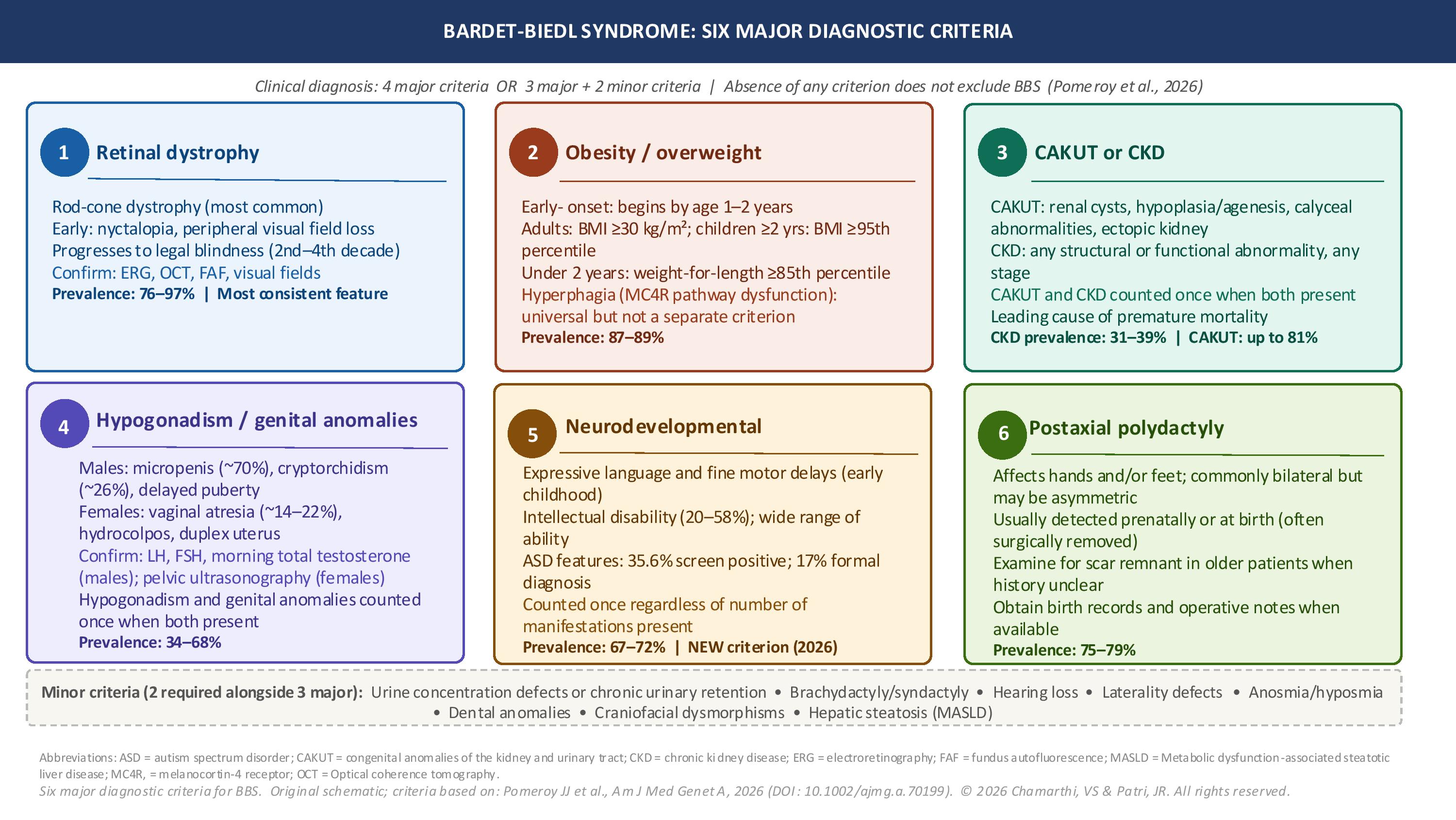
Diagnostic Criteria for Bardet-Biedl Syndrome. Summary of the 6 major diagnostic criteria for Bardet-Biedl syndrome, including retinal dystrophy, obesity/overweight, congenital anomalies of the kidney and urinary tract (CAKUT) or chronic kidney disease (CKD), hypogonadism/genital anomalies, neurodevelopmental abnormalities, and postaxial polydactyly. This figure includes prevalence estimates, key clinical features, and associated minor diagnostic criteria.
Contributed by V Sushma Chamarthi, MD, and JR Patri, MD
References
Melluso A, Secondulfo F, Capolongo G, Capasso G, Zacchia M. Bardet-Biedl Syndrome: Current Perspectives and Clinical Outlook. Therapeutics and clinical risk management. 2023:19():115-132. doi: 10.2147/TCRM.S338653. Epub 2023 Jan 30 [PubMed PMID: 36741589]
Level 3 (low-level) evidencePomeroy JJ, Richards J, Sweeney BR, Kumar S, Queen KE, Zaritsky J, Cramer CH, Traboulsi EI, Scruggs BA, Davis EE, Keifer E, McGibbon E, Ogden T, De Graaf B, Hymers T, Forsythe E, Beales P. Streamlining Diagnosis of Bardet-Biedl Syndrome: New Diagnostic Algorithm With Updated Criteria. American journal of medical genetics. Part A. 2026 May 23:():. doi: 10.1002/ajmg.a.70199. Epub 2026 May 23 [PubMed PMID: 42175648]
Dollfus H, Lilien MR, Maffei P, Verloes A, Muller J, Bacci GM, Cetiner M, van den Akker ELT, Grudzinska Pechhacker M, Testa F, Lacombe D, Stokman MF, Simonelli F, Gouronc A, Gavard A, van Haelst MM, Koenig J, Rossignol S, Bergmann C, Zacchia M, Leroy BP, Mosbah H, Van Eerde AM, Mekahli D, Servais A, Poitou C, Valverde D. Bardet-Biedl syndrome improved diagnosis criteria and management: Inter European Reference Networks consensus statement and recommendations. European journal of human genetics : EJHG. 2024 Nov:32(11):1347-1360. doi: 10.1038/s41431-024-01634-7. Epub 2024 Jul 31 [PubMed PMID: 39085583]
Level 3 (low-level) evidenceHaqq AM, Chung WK, Dollfus H, Haws RM, Martos-Moreno GÁ, Poitou C, Yanovski JA, Mittleman RS, Yuan G, Forsythe E, Clément K, Argente J. Efficacy and safety of setmelanotide, a melanocortin-4 receptor agonist, in patients with Bardet-Biedl syndrome and Alström syndrome: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial with an open-label period. The lancet. Diabetes & endocrinology. 2022 Dec:10(12):859-868. doi: 10.1016/S2213-8587(22)00277-7. Epub 2022 Nov 7 [PubMed PMID: 36356613]
Level 1 (high-level) evidenceTomlinson JW. Bardet-Biedl syndrome: A focus on genetics, mechanisms and metabolic dysfunction. Diabetes, obesity & metabolism. 2024 Apr:26 Suppl 2():13-24. doi: 10.1111/dom.15480. Epub 2024 Feb 1 [PubMed PMID: 38302651]
Solarat C, Valverde D. Clinical and molecular diagnosis of Bardet-Biedl syndrome (BBS). Methods in cell biology. 2023:176():125-137. doi: 10.1016/bs.mcb.2022.12.014. Epub 2023 Jan 9 [PubMed PMID: 37164534]
Beales PL, Cetiner M, Haqq AM, Miller J, Shoemaker AH, Valverde D, Zacchia M, Dollfus H. Hyperphagia in Bardet-Biedl syndrome: Pathophysiology, burden, and management. Obesity reviews : an official journal of the International Association for the Study of Obesity. 2025 Jul:26(7):e13915. doi: 10.1111/obr.13915. Epub 2025 Apr 4 [PubMed PMID: 40186386]
Finkelberg I, Polichronidou IM, Hühne T, Brensing P, Karaterzi S, Jaegers J, Gäckler A, Pape L, Cetiner M. Patient and caregiver experiences with a patient-support program for setmelanotide treatment of patients with Bardet-Biedl syndrome. Orphanet journal of rare diseases. 2025 Jun 8:20(1):290. doi: 10.1186/s13023-025-03835-9. Epub 2025 Jun 8 [PubMed PMID: 40484968]
Lazareva J, Brady SM, Yanovski JA. An evaluation of setmelanotide injection for chronic weight management in adult and pediatric patients with obesity due to Bardet-Biedl syndrome. Expert opinion on pharmacotherapy. 2023 Apr:24(6):667-674. doi: 10.1080/14656566.2023.2199152. Epub 2023 Apr 6 [PubMed PMID: 37013719]
Level 3 (low-level) evidenceNowak-Ciołek M, Ciołek M, Tomaszewska A, Hildebrandt F, Kitzler T, Deutsch K, Lemberg K, Shril S, Szczepańska M, Zachurzok A. Collaborative effort: managing Bardet-Biedl syndrome in pediatric patients. Case series and a literature review. Frontiers in endocrinology. 2024:15():1424819. doi: 10.3389/fendo.2024.1424819. Epub 2024 Jul 18 [PubMed PMID: 39092285]
Level 2 (mid-level) evidence