Introduction
Arginase deficiency (argininemia) is an autosomal recessive metabolic disorder characterized by arginine accumulation and variable hyperammonemia. The disorder typically presents initially with slow growth, followed by developmental delay and cognitive regression. Ammonia levels may vary according to the patient's age and clinical status. Without appropriate treatment, arginase deficiency may lead to progressive neurologic regression. Newborn screening often identifies affected newborns by detecting plasma arginine levels approximately 3 to 4 times the upper limit of the reference range. However, the characteristic progressively debilitating neurologic manifestations typically develop between 2 and 4 years of age. Hyperargininemia is the characteristic biochemical finding that distinguishes arginase deficiency from other urea cycle disorders. Treatment is similar to that of other classic urea cycle disorders, although hyperammonemia may be mild or absent. When present, hyperammonemia responds adequately to ammonia-reducing interventions. Chronic treatment consists of protein restriction and nitrogen-scavenging medications.[1][2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Biallelic pathogenic or likely pathogenic variants in the ARG1 gene lead to deficiency or instability of the arginase 1 enzyme. The ARG1 gene is located on chromosome 6q23.2. The product of this gene, the arginase 1 enzyme, is found primarily in the hepatic cytosol and is responsible for the final step of the urea cycle: the hydrolysis of L-arginine into urea and ornithine.[3] In arginase deficiency, this defect prevents the proper breakdown of arginine, leading to its systemic accumulation. A second gene, ARG2, encodes a different arginase enzyme (arginase 2) that is found mainly in the mitochondria of extrahepatic tissues, such as the kidney, brain, and gastrointestinal tract. However, arginase 2 expression is insufficient, and the enzyme is located in a different cellular compartment, preventing compensation for the primary defect in the hepatic urea cycle.[4]
Epidemiology
Arginase deficiency is the rarest of the urea cycle disorders, with an estimated incidence ranging from approximately 1 in 300,000 to 1 in 1,000,000 births.[5] More than 250 cases have been reported in the literature to date. While a precise genotype-phenotype correlation has not been fully established, disease severity often appears to correlate with the nature of the biallelic ARG1 variants. For instance, homozygous or compound heterozygous loss-of-function variants are typically associated with a more severe, classic phenotype than are some missense variants. The disorder occurs in all racial and ethnic groups and does not appear to have a higher prevalence in any specific population.[1][6][7]
Pathophysiology
Arginase 1, encoded by the ARG1 gene and found primarily in the liver and erythrocytes, catalyzes the fifth and final reaction of the urea cycle: the hydrolysis of L-arginine into ornithine and urea (see Image. Urea Cycle Disorder: Arginase Deficiency). A deficiency or absence of arginase 1 activity blocks this final step, causing the systemic accumulation of arginine (hyperargininemia) in the blood and central nervous system. Arginine accumulation begins at birth. Unlike proximal urea cycle disorders, significant hyperammonemia is often absent. The primary pathophysiology is driven by the neurotoxicity of arginine itself, which is shunted into alternative metabolic pathways. Alternative arginine metabolism generates toxic metabolites, such as guanidino compounds, which are believed to be responsible for the characteristic progressive neurologic symptoms (eg, spasticity and cognitive delay) that typically become clinically apparent around 1 to 3 years of age.[8]
A characteristic biochemical finding is the accumulation of orotic acid (orotic aciduria). Orotic aciduria occurs because the block in arginine hydrolysis leads to a relative deficiency of ornithine. Ornithine is required for ornithine transcarbamylase to function. Reduced ornithine availability causes an accumulation of carbamoyl phosphate, which is subsequently shunted from the urea cycle into the pyrimidine synthesis pathway, resulting in orotic acid overproduction.[6][9]
Toxicokinetics
The toxicokinetics of argininemia are distinct from those of other urea cycle disorders and are not primarily driven by ammonia accumulation. The central mechanism is the neurotoxicity of chronic hyperargininemia and its metabolic byproducts. Extremely high cellular arginine levels saturate alternative enzyme pathways, most notably arginine-glycine amidinotransferase. Shunting through this pathway leads to the overproduction of guanidino compounds.[2] Guanidino compounds are believed to be the primary neurotoxins responsible for the characteristic progressive spasticity and cognitive decline.[10] Furthermore, excess arginine dysregulates the nitric oxide pathway by overwhelming nitric oxide synthase. Dysregulation of the nitric oxide pathway can contribute to endothelial dysfunction and has been proposed as another mechanism of neurotoxicity.[8] Although chronic, progressive cerebral injury (including cortical atrophy in the parietal, occipital, and frontal lobes) is linked to these toxic metabolites, acute hyperammonemia can still occur, particularly during catabolic states (eg, illness).[11] A sudden increase in the ammonia level can cause cerebral edema, but acute hyperammonemia is an intermittent event and does not account for the characteristic progressive course of the disease.
History and Physical
Arginase deficiency (argininemia) rarely presents during the neonatal period or infancy with the life-threatening hyperammonemia characteristic of proximal urea cycle disorders. The classic presentation is a progressive neurologic syndrome that typically becomes clinically apparent between 1 and 3 years of age. The earliest signs are often slowed linear growth, followed by the hallmark finding of the condition: progressive spastic diplegia (abnormal muscle tightening and stiffness, primarily in the lower extremities). Progressive spastic diplegia is accompanied by a plateau or regression of cognitive development and the loss of previously acquired motor milestones.[3] Because the primary clinical picture is progressive spasticity, often with only mild or intermittent hyperammonemia, the condition is frequently misdiagnosed as cerebral palsy or hereditary spastic paraplegia. Progressive spasticity occurs in nearly 80% to 90% of affected individuals and is the most consistent neurologic finding on physical examination.
As the disease progresses, especially if undiagnosed or inadequately treated, patients develop severe complications.[12] Complications include worsening spasticity leading to loss of ambulation, intellectual disability, seizures, and behavioral issues such as attention-deficit/hyperactivity disorder and aggression.[13] Nearly half to two-thirds of the affected individuals manifest with seizures, and generalized tonic-clonic seizures are the most common type. Intermittent episodic hyperammonemia can still occur, especially when induced by catabolic stressors (eg, infections), high protein intake, or certain medications (eg, valproate), but hyperammonemia does not cause the chronic, progressive neurologic decline.[13] Extraneurological manifestations are less common but can include liver dysfunction (usually mild, although fibrosis or cirrhosis have occasionally been reported) and bone involvement.[3]
Evaluation
The evaluation for arginase deficiency is typically initiated after positive newborn screening results or, increasingly, in a symptomatic child not identified through screening.
Newborn Screening
Newborn screening programs screen for hyperargininemia by quantifying arginine levels in dried blood spots. However, not all screening panels include arginase deficiency. Furthermore, false-negative results are known to occur because arginine levels may be only mildly elevated during the neonatal period before significant protein catabolism begins.[2][5]
Symptomatic Presentation
A high index of suspicion is warranted for any child presenting with progressive spastic diplegia, developmental regression, or seizures, especially when cognitive decline has been noted.[13][14]
Biochemical Diagnosis
The essential diagnostic evaluation for a suspected case includes:
- Plasma amino acid analysis: This is the most critical test. Results reveal an isolated elevation in the plasma arginine level, often 4 to 10 times the upper limit of the reference range.
- Urine organic acid analysis: Results typically show an elevated orotic acid level (orotic aciduria). Orotic aciduria is a key finding that differentiates arginase deficiency from other diagnoses and confirms a block in the urea cycle.
- Plasma ammonia level: The plasma ammonia level provides supportive diagnostic information. Results may be within the reference range, mildly elevated, or significantly elevated (eg, greater than 200 µg/dL), depending on the patient's current metabolic (catabolic) state. An ammonia level within the reference range does not rule out argininemia.
Neuroimaging
Neuroimaging findings in the central nervous system result directly from chronic neurotoxicity caused by elevated plasma arginine levels and guanidino compound metabolites. Common findings include cortical atrophy, particularly in the frontal and parietal lobes, ischemic changes, and edema. Neuroimaging findings may resemble the cerebral injury seen in patients affected by hypoxic-ischemic events. The similarity between these findings is a primary reason the clinical presentation is frequently misdiagnosed as cerebral palsy.[11]
Confirmatory Testing
The diagnosis is confirmed by molecular genetic testing of ARG1 to identify biallelic pathogenic variants. Molecular genetic testing is now the standard confirmatory step.[5] Less commonly, an enzyme assay on red blood cells can be performed, with results showing arginase 1 activity of less than 1%. More than 98% of cases with confirmed molecular genetic analysis have sequence variants, and less than 2% may have copy number variants, so the molecular genetic test should be planned accordingly. The genotype-phenotype correlation in arginase deficiency is related to the residual enzyme activity associated with the variant.
Critical Diagnostic Warning
The initial evaluation is critical. Argininemia is a distal urea cycle disorder in which arginine is the primary toxin. In contrast, proximal urea cycle disorders, such as ornithine transcarbamylase deficiency, are treated with supplemental arginine to promote nitrogen excretion. Misdiagnosing argininemia as a proximal urea cycle disorder and administering arginine can cause severe, rapid neurologic harm.
Treatment / Management
Treatment of arginase deficiency is lifelong and focuses on 2 distinct clinical scenarios: acute hyperammonemic crises and the more common progressive chronic disease.
Management of Acute Hyperammonemia
Acute hyperammonemic crises are less frequent than those associated with other urea cycle disorders but are medical emergencies. Treatment includes:
- Protein cessation: All protein intake should be stopped for 24 to 48 hours.
- Intravenous fluids and calories: Provide high-calorie, protein-free sources (eg, 10% intravenous dextrose) to promote anabolism and reverse catabolism.
- Nitrogen scavengers: Intravenous nitrogen-scavenging medications, such as sodium phenylacetate or sodium benzoate, are used to treat moderate or severe hyperammonemia.
- Dialysis: In cases of severe hyperammonemia refractory to medical interventions, hemodialysis or continuous renal replacement therapy is highly effective.[13] (B3)
Long-Term Management
The primary goal of chronic therapy is to control plasma arginine levels to prevent progressive, irreversible neurotoxicity.[8]
Foundational therapy: The traditional approach aims to limit arginine intake and provide alternative pathways for the excretion of nitrogen waste.
- Diet: Lifelong dietary protein restriction is required. A specialized arginine-free essential amino acid formula supplements the diet to prevent malnutrition.
- Nitrogen scavengers: Daily maintenance dosing of nitrogen scavengers (eg, sodium phenylbutyrate or glycerol phenylbutyrate) is used.
- Limitations of the standard of care: Results from recent natural history studies indicate that foundational therapy is often insufficient.[12] Many patients do not maintain target arginine levels, and the highly restrictive diet creates a significant treatment burden. Most importantly, foundational therapy has not been shown to reliably halt the progression of neurologic symptoms, such as spasticity, in many patients. (A1)
Enzyme replacement therapy: The most significant advancement in treatment is the development of enzyme replacement therapy, which directly targets elevated arginine levels, the underlying metabolic abnormality.
- Drug: Pegzilarginase-nbln is a recombinant human arginase 1 enzyme administered via subcutaneous injection.
- Mechanism: Pegzilarginase-nbln provides an exogenous source of arginase 1 enzyme activity and lowers plasma arginine levels by converting arginine to urea and ornithine.
- Efficacy: Results from the phase 3 Pegzilarginase Effect on Arginase 1 Deficiency Clinical Endpoints (PEACE) trial demonstrated rapid, significant, and sustained reductions in plasma arginine levels.[15] This metabolic control was directly associated with statistically significant improvements in mobility and functional outcomes compared to placebo. Motor function outcomes improved with pegzilarginase-nbln, although the difference compared with placebo was not statistically significant. Pegzilarginase-nbln received accelerated approval from the US Food and Drug Administration on February 23, 2026, for the treatment of hyperargininemia in adults and children aged 2 years or older with arginase 1 deficiency, in conjunction with dietary protein restriction. The accelerated approval was based on reduced plasma arginine levels, and continued approval may depend on confirmation of clinical benefit. [FDA. Loargys Prescribing Information] Pegzilarginase was previously authorized in the European Union in December 2023 under exceptional circumstances and additional monitoring.[EMA. Loargys] (A1)
Supportive Care and Other Treatment
Supportive care: Physical and occupational therapy are critical for managing spasticity and maintaining mobility.
Developmental support: Care from a developmental pediatrician and an interdisciplinary team is paramount for nearly all affected individuals from an early age. An individualized education plan is needed for most affected individuals to maximize independence and productivity later in life.
Seizure treatment: Seizures are common and treated with standard antiseizure medications, such as phenobarbital or carbamazepine. Valproate (valproic acid) is strongly contraindicated because it interferes with the urea cycle and can induce hyperammonemia.[13](B3)
Liver transplant: Individuals with chronic end-stage liver disease may require a liver transplant. Liver transplant most closely approximates curative therapy because it provides a fully functional liver. However, the significant associated risks must be carefully weighed against the benefits of newer medical therapies. Moreover, direct evidence demonstrating its effects on elevated arginine levels and neurologic status is lacking.
Monitoring: Lifelong, regular monitoring by a metabolic specialist and a dietitian is essential. Monitoring includes frequent measurement of plasma amino acid levels (especially arginine), plasma ammonia levels, and liver function.
Investigational Therapies
Research into one-time, curative treatments continues. Results from preclinical studies of messenger RNA-based therapy, gene therapy using adeno-associated virus vectors, and clustered regularly interspaced short palindromic repeats-based genome editing to correct the gene in hepatocytes showed promise, but these therapies remain investigational.[5][9][16][17]
Differential Diagnosis
A differential diagnosis must address 2 domains: the clinical presentation and the biochemical profile.
Clinical and Neurological Differential
This is the most common pitfall, leading to significant diagnostic delays.
- Cerebral palsy: Argininemia is most frequently misdiagnosed as cerebral palsy.[14] The key differentiator between cerebral palsy and arginase deficiency is progression: cerebral palsy is a nonprogressive motor disorder, whereas the symptoms of argininemia will progressively worsen over time.
- Hereditary spastic paraplegia: This group of genetic disorders also causes progressive lower-extremity spasticity. Argininemia is now considered a treatable form of spastic paraplegia and should be excluded through plasma amino acid analysis during the evaluation of hereditary spastic paraplegia.
Biochemical Differential
Proximal urea cycle disorders (eg, ornithine transcarbamylase deficiency and carbamoyl phosphate synthetase 1 deficiency): These disorders are biochemically distinct. They present with severe, life-threatening hyperammonemia during the neonatal period and low to reference-range plasma arginine levels. In contrast, argininemia presents with significantly elevated plasma arginine levels and often reference-range or only mildly elevated ammonia levels.
Lysinuric protein intolerance: This rare autosomal recessive disorder is caused by variants in the SLC7A7 gene. Lysinuric protein intolerance can also present with hyperammonemia and elevated plasma arginine levels, mimicking argininemia on newborn screening.[9] The key differentiator is that lysinuric protein intolerance is an amino acid transport disorder characterized by elevated urinary levels of lysine, ornithine, and arginine (lysinuria), as well as low plasma levels of these amino acids.
Prognosis
The prognosis for argininemia is variable but predictable, depending directly on 2 factors: the age at diagnosis and the success of long-term metabolic control. Historically, the prognosis was poor because foundational therapy alone was often inadequate. Results from natural history studies showed that treatment with a protein-restricted diet and nitrogen-scavenging medications was frequently insufficient to control plasma arginine levels.[12] As a result, most patients experienced progressive, irreversible neurologic damage, including severe spasticity, loss of ambulation, and moderate to severe intellectual disability, leading to reduced quality of life.
The prognosis may improve with arginine-lowering enzyme replacement therapy. Results from the PEACE trial demonstrated that pegzilarginase significantly reduced plasma arginine levels, with mobility outcomes favoring treatment.[15] Results from a recent extension study showed that pegzilarginase improved biochemical and clinical outcomes. Temporary treatment discontinuation was associated with worsening outcomes, whereas treatment resumption was associated with subsequent improvement in clinical and biochemical markers.[18]
The key prognostic factor remains the irreversibility of existing neurologic damage.[2] Patients diagnosed late (eg, after a misdiagnosis of cerebral palsy) and treated with enzyme replacement therapy may experience stabilization of disease progression and recovery of some lost motor or cognitive milestones. The potential for irreversible injury underscores the critical importance of early diagnosis and timely, effective treatment.
Complications
The primary complications of argininemia result from chronic, irreversible neurologic damage caused by hyperargininemia and related metabolites. If the diagnosis is delayed or treatment does not adequately control plasma arginine levels, patients may develop:
- Progressive spastic diplegia, leading to joint contractures and loss of ambulation
- Seizures
- Moderate-to-severe intellectual disability
- Cognitive and behavioral deficits
- Short stature
- Hepatic complications: While neurological symptoms predominate, the liver is the primary site of the enzyme defect. Late manifestations, reflecting long-term metabolic stress, can include the development of hepatic fibrosis, cirrhosis, and, in rare cases, hepatocellular carcinoma.[19]
Results from natural history studies showed that these complications can develop even in patients adhering to standard-of-care dietary and nitrogen-scavenging therapy.[12] Although many infants are now identified through newborn screening, screening is not universal, and false-negative results can occur. Delayed diagnosis, often due to a misdiagnosis of cerebral palsy, is a major complication that contributes to poor prognosis.
Consultations
Consultations for patients with hyperarginemia include:
- Pediatric neurologist
- Medical geneticist
- Physical therapist
- Laboratory clinician
- Nutritionist
Deterrence and Patient Education
Effective prevention of complications relies on early diagnosis and rigorous, lifelong treatment. Patient and family education is critical and should focus on:
- The goal of treatment: The primary goal is to keep plasma arginine levels as low as possible. Families must understand that although ammonia levels are monitored, chronic neurologic damage (spasticity and cognitive decline) is caused by elevated arginine levels.
- Treatment adherence: Families must understand the critical importance of adhering to the demanding low-protein diet, nitrogen-scavenging medications, and enzyme replacement therapy. Failure to control arginine levels can lead to progressive, irreversible neurologic damage.
- Recognizing misdiagnosis: Families and clinicians should be educated that argininemia can mimic cerebral palsy. Any diagnosis of cerebral palsy associated with progressive symptoms should prompt immediate evaluation for argininemia.
- Metabolic crises: Families must be able to recognize the early signs of a catabolic state (eg, fever, vomiting, and lethargy) that could trigger hyperammonemia and know when to seek emergency care.
- Genetic counseling: Genetic counseling is essential for affected individuals and families to discuss the autosomal recessive inheritance pattern and the risk to future pregnancies.
Pearls and Other Issues
Pearls regarding arginase deficiency include:
- The single most important clinical pearl is that argininemia is frequently misdiagnosed as cerebral palsy or hereditary spastic paraplegia.
- Argininemia should be considered a treatable form of spastic paraplegia and should be excluded in any child with progressive spasticity, even if the ammonia level is within the reference range.
- The primary neurotoxin is arginine, not ammonia. Treatment and prognosis are dictated by the success of lowering plasma arginine levels.
- Valproic acid is contraindicated for seizures because it can induce hyperammonemia.
- A plasma ammonia level within the reference range does not rule out the diagnosis.
- Arginase deficiency is currently listed as a secondary condition on the Recommended Uniform Screening Panel. Therefore, not all states may include arginase deficiency in newborn screening.
- Newborn screening can produce false-negative results. Newborn screening results within the reference range do not rule out argininemia in a symptomatic child.
- Misdiagnosing argininemia as a proximal urea cycle disorder, which may be treated with arginine, is a critical and dangerous error.
- Prenatal testing: For families with a known pathogenic variant, prenatal diagnosis is available through gene sequencing of samples obtained by chorionic villus sampling or amniocentesis. Enzyme activity assays of amniocytes are unreliable and should not be used.
Enhancing Healthcare Team Outcomes
Effective treatment of argininemia, a condition often misdiagnosed as cerebral palsy, demands a paradigm shift in interprofessional strategy and communication. The team's primary ethical responsibility is to coordinate care focused on controlling neurotoxic arginine levels, not just intermittent hyperammonemia. Effective care requires seamless communication among a pediatric neurologist, a medical geneticist, a metabolic dietitian, a laboratory clinician, and a primary care clinician.
The interprofessional team should ensure timely diagnosis and avoid irreversible neurologic harm caused by diagnostic delays. Pharmacists must collaborate on dosing of nitrogen-scavenging medications and newer therapies, while metabolic dietitians develop individualized restrictive diets. Nurses and physical therapists are essential for monitoring adherence, treating spasticity, and enhancing patient safety. Integrated care coordination among specialists in genetics, neurology, and nutrition is essential to enhance team performance, reduce the risk of progressive neurologic decline, and optimize patient-centered outcomes.
Media
(Click Image to Enlarge)
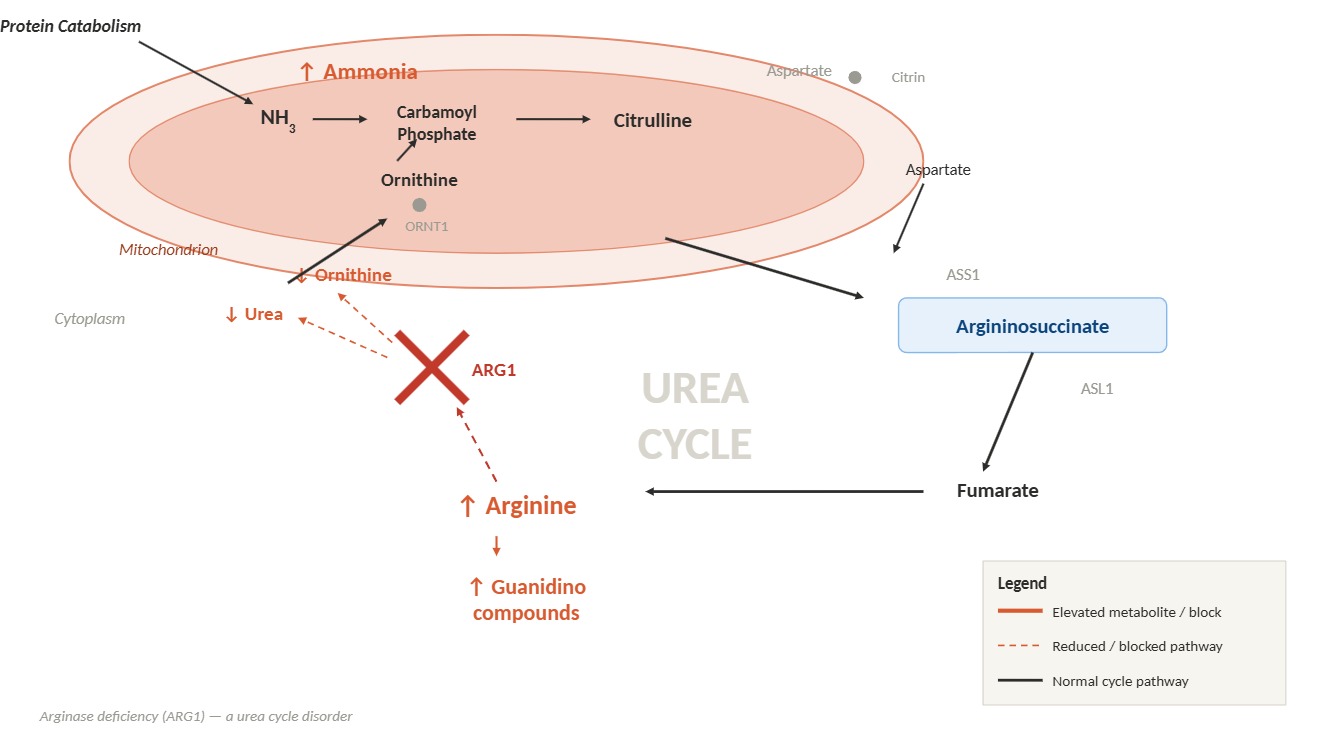
Urea Cycle Disorder: Arginase Deficiency. A block in the enzymatic conversion of arginine to urea and ornithine results in hyperargininemia, hyperammonemia, and impaired urea cycle function.
Contributed by L Sharma, MD
References
Morris SM Jr. Arginases and arginine deficiency syndromes. Current opinion in clinical nutrition and metabolic care. 2012 Jan:15(1):64-70. doi: 10.1097/MCO.0b013e32834d1a08. Epub [PubMed PMID: 22037011]
Level 3 (low-level) evidenceAdam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Sun A, Crombez EA, Wong D. Arginase Deficiency. GeneReviews(®). 1993:(): [PubMed PMID: 20301338]
Sin YY, Baron G, Schulze A, Funk CD. Arginase-1 deficiency. Journal of molecular medicine (Berlin, Germany). 2015 Dec:93(12):1287-96. doi: 10.1007/s00109-015-1354-3. Epub 2015 Oct 14 [PubMed PMID: 26467175]
Waisbren SE, Gropman AL, Members of the Urea Cycle Disorders Consortium (UCDC), Batshaw ML. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. Journal of inherited metabolic disease. 2016 Jul:39(4):573-84. doi: 10.1007/s10545-016-9942-0. Epub 2016 May 23 [PubMed PMID: 27215558]
Therrell BL, Currier R, Lapidus D, Grimm M, Cederbaum SD. Newborn screening for hyperargininemia due to arginase 1 deficiency. Molecular genetics and metabolism. 2017 Aug:121(4):308-313. doi: 10.1016/j.ymgme.2017.06.003. Epub 2017 Jun 20 [PubMed PMID: 28659245]
Iyer R, Jenkinson CP, Vockley JG, Kern RM, Grody WW, Cederbaum S. The human arginases and arginase deficiency. Journal of inherited metabolic disease. 1998:21 Suppl 1():86-100 [PubMed PMID: 9686347]
Level 3 (low-level) evidenceDiez-Fernandez C, Rüfenacht V, Gemperle C, Fingerhut R, Häberle J. Mutations and common variants in the human arginase 1 (ARG1) gene: Impact on patients, diagnostics, and protein structure considerations. Human mutation. 2018 Aug:39(8):1029-1050. doi: 10.1002/humu.23545. Epub 2018 Jun 21 [PubMed PMID: 29726057]
Diaz GA, Bechter M, Cederbaum SD. The role and control of arginine levels in arginase 1 deficiency. Journal of inherited metabolic disease. 2023 Jan:46(1):3-14. doi: 10.1002/jimd.12564. Epub 2022 Oct 13 [PubMed PMID: 36175366]
Yahyaoui R, Blasco-Alonso J, Benito C, Rodríguez-García E, Andrade F, Aldámiz-Echevarría L, Muñoz-Hernández MC, Vega AI, Pérez-Cerdá C, García-Martín ML, Pérez B. A new metabolic disorder in human cationic amino acid transporter-2 that mimics arginase 1 deficiency in newborn screening. Journal of inherited metabolic disease. 2019 May:42(3):407-413. doi: 10.1002/jimd.12063. Epub 2019 Feb 21 [PubMed PMID: 30671984]
Deignan JL, Marescau B, Livesay JC, Iyer RK, De Deyn PP, Cederbaum SD, Grody WW. Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia. Molecular genetics and metabolism. 2008 Feb:93(2):172-8 [PubMed PMID: 17997338]
Level 3 (low-level) evidenceGüngör S, Akinci A, Firat AK, Tabel Y, Alkan A. Neuroimaging findings in hyperargininemia. Journal of neuroimaging : official journal of the American Society of Neuroimaging. 2008 Oct:18(4):457-62. doi: 10.1111/j.1552-6569.2007.00217.x. Epub 2008 Jan 7 [PubMed PMID: 18321250]
Level 3 (low-level) evidenceBin Sawad A, Pothukuchy A, Badeaux M, Hodson V, Bubb G, Lindsley K, Uyei J, Diaz GA. Natural history of arginase 1 deficiency and the unmet needs of patients: A systematic review of case reports. JIMD reports. 2022 Jul:63(4):330-340. doi: 10.1002/jmd2.12283. Epub 2022 Mar 25 [PubMed PMID: 35822089]
Level 1 (high-level) evidenceCederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Molecular genetics and metabolism. 2004 Apr:81 Suppl 1():S38-44 [PubMed PMID: 15050972]
Level 3 (low-level) evidenceBurlina A, Ardissone A, Battini R, Burlina A, Gasperini S, Pession A, Porta F, Vici CD. Arginase 1 deficiency: a treatable form of spastic paraplegia. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2025 Sep:46(9):4219-4228. doi: 10.1007/s10072-025-08153-3. Epub 2025 Apr 16 [PubMed PMID: 40237972]
Russo RS, Gasperini S, Bubb G, Neuman L, Sloan LS, Diaz GA, Enns GM, PEACE Investigators. Efficacy and safety of pegzilarginase in arginase 1 deficiency (PEACE): a phase 3, randomized, double-blind, placebo-controlled, multi-centre trial. EClinicalMedicine. 2024 Feb:68():102405. doi: 10.1016/j.eclinm.2023.102405. Epub 2024 Jan 12 [PubMed PMID: 38292042]
Level 1 (high-level) evidenceSin YY, Price PR, Ballantyne LL, Funk CD. Proof-of-Concept Gene Editing for the Murine Model of Inducible Arginase-1 Deficiency. Scientific reports. 2017 May 31:7(1):2585. doi: 10.1038/s41598-017-02927-2. Epub 2017 May 31 [PubMed PMID: 28566761]
Asrani KH, Cheng L, Cheng CJ, Subramanian RR. Arginase I mRNA therapy - a novel approach to rescue arginase 1 enzyme deficiency. RNA biology. 2018:15(7):914-922. doi: 10.1080/15476286.2018.1475178. Epub 2018 Jul 24 [PubMed PMID: 29923457]
Faraguna MC, Crescitelli V, Pretese R, Bolgè MV, Marchetti V, Sgroi G, Sala S, Gigante S, Bonfanti C, Balduzzi A, Gasperini S. Pegzilarginase in Arginase 1 Deficiency: Clinical and Biochemical Effects of Treatment Initiation, Discontinuation and Re-Initiation. Children (Basel, Switzerland). 2026 Apr 28:13(5):. doi: 10.3390/children13050610. Epub 2026 Apr 28 [PubMed PMID: 42194135]
Lal V, Khera D, Gupta G, Singh K, Sharma P. A Case of Hyperargininaemia Presenting at Unusually Low Age. Journal of clinical and diagnostic research : JCDR. 2017 Jul:11(7):BD01-BD03. doi: 10.7860/JCDR/2017/29270.10263. Epub 2017 Jul 1 [PubMed PMID: 28892883]
Level 3 (low-level) evidence