Introduction
Anemia is characterized by a reduction in the circulating red blood cell (RBC) mass, reflected by decreased hemoglobin concentration, hematocrit, or RBC count. The 2024 World Health Organization (WHO) guideline on hemoglobin cutoffs to define anemia represents the first significant update since the original 1968 thresholds, reaffirming most longstanding values while introducing updated cutoffs for specific subgroups.[1][WHO. Guideline on Haemoglobin Cutoffs to Define Anaemia in Individuals and Populations] The thresholds are summarized as follows:
- Adult men: Hemoglobin < 13.0 g/dL
- Adult nonpregnant women: Hemoglobin < 12.0 g/dL
- Pregnant women: Hemoglobulin < 11.0 g/dL (first and third trimesters); hemoglobin < 10.5 g/dL (second trimester, updated 2024)
- Children aged 6 to 23 months: Hemoglobulin < 10.5 g/dL (updated 2024, previously 11.0 g/dL)
- Children aged 2 to 11 years: Hemoglobin < 11.0 g/dL
- Children aged 12 to 14 years: Hemoglobin < 12.0 g/dL
Erythropoietin (EPO), produced predominantly by peritubular interstitial fibroblasts in the kidney, serves as the principal hormonal regulator of erythropoiesis. Tissue-level hypoxia constitutes the primary physiologic stimulus for EPO secretion, mediated through hypoxia-inducible factor 2α. Circulating EPO concentrations are generally inversely related to hemoglobin levels; as hemoglobin falls, EPO levels rise exponentially, thereby stimulating erythroid progenitor survival, proliferation, and differentiation.[2] In chronic kidney disease (CKD), progressive renal parenchymal loss blunts the compensatory EPO response, resulting in a relative EPO deficiency that substantially contributes to CKD-associated anemia. Similarly, in anemia of inflammation, also termed anemia of chronic disease, EPO levels are typically elevated above the reference range but remain inappropriately low relative to the degree of anemia, a phenomenon attributed to the inhibitory effects of inflammatory cytokines on EPO gene expression.[3]
This anemia overview article serves as a hub for the anemia topic section, providing a systematic framework for clinicians encountering anemia across diverse clinical settings, including primary care, hospital medicine, perioperative care, and obstetrics. The article directs readers to dedicated articles for in-depth discussion of individual anemia subtypes, including iron deficiency anemia, vitamin B12 and folate deficiency, hemolytic anemias, sickle cell disease, thalassemia syndromes, myelodysplastic syndromes, aplastic anemia, and anemia of chronic kidney disease.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
A practical approach begins by determining whether anemia is associated with an appropriate marrow response. Clinicians can assess the marrow response using the reticulocyte production index (RPI).[4][5] An RPI less than 2 generally suggests an inadequate marrow response, whereas an RPI greater than 3 supports an appropriate marrow response. Intermediate values should be interpreted in the clinical context.[NIH. Red Cell Manual] The mean corpuscular volume (MCV) further classifies hypoproliferative anemias into microcytic, normocytic, and macrocytic categories.[6]
Hypoproliferative Microcytic Anemia (MCV < 80 fL)
Iron deficiency anemia is the most common cause of anemia worldwide, accounting for approximately 66% of all cases (see Image. Normal Versus Iron Deficiency Anemia ).[7] The disorder results from depleted iron stores due to inadequate dietary intake, impaired absorption (celiac disease, Helicobacter pylori infection, postbariatric surgical procedures), increased physiologic demand (pregnancy, growth), or chronic blood loss (gastrointestinal tract or menstrual). Additional microcytic etiologies include:
- Anemia of inflammation or anemia of chronic disease: These disorders may also present with normocytic findings and result from hepcidin-mediated iron sequestration in settings of chronic immune activation.[3]
- Thalassemia syndromes: These inherited disorders of globin chain synthesis cause ineffective erythropoiesis; α-thalassemia results from HBA gene deletions, whereas β-thalassemia results from HBB gene mutations.[8]
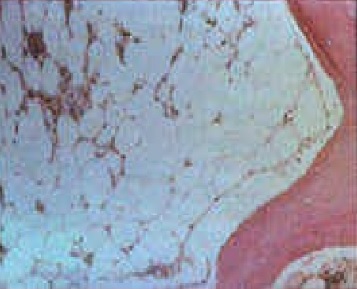
- Sideroblastic anemia: These congenital or acquired disorders of heme biosynthesis are characterized by ring sideroblasts in bone marrow and may produce a dimorphic microcytic and macrocytic red cell population (see Image. Sideroblastic Anemia).[9]
- Lead poisoning: Lead poisoning inhibits δ-aminolevulinic acid dehydratase and ferrochelatase, causing impaired heme synthesis, and peripheral smear findings typically show basophilic stippling.[10]
Please see StatPearls' companion references, "Iron Deficiency Anemia," "Thalassemia," and "Sideroblastic Anemia," for further information.
Hypoproliferative Normocytic Anemia (MCV 80 to 100 fL)
Normocytic anemias encompass a broad differential. Anemia of inflammation is the most common cause of normocytic anemia in hospitalized patients.[11] Other important etiologies include:
- Anemia of chronic kidney disease: This disease results from inadequate EPO production, uremic inhibition of erythropoiesis, and shortened RBC lifespan.[11]
- Aplastic anemia: This immune-mediated disorder involves the destruction of hematopoietic stem cells, leading to pancytopenia and hypocellular bone marrow.[12] (see Image. Aplastic Anemia Bone Marrow).
- Pure red cell aplasia: This condition results from selective failure of erythroid precursors and is associated with thymoma, parvovirus B19, and certain medications.[13]
- Myelophthisic anemia: This disorder results from marrow infiltration by fibrosis (myelofibrosis), granulomatous disease, or a metastatic malignant neoplasm; peripheral smear findings show a characteristic leukoerythroblastic picture with nucleated red blood cells and immature myeloid cells. Please see StatPearls' companion reference, "Myelophthisic Anemia," for further information.
- Multiple myeloma and plasma cell dyscrasias: Anemia results from marrow infiltration, cytokine-mediated suppression of erythropoiesis, relative erythropoietin deficiency due to renal impairment, and chemotherapy-related myelosuppression.[14]
Mixed nutritional deficiencies: For example, iron and vitamin B12 deficiencies occurring together can result in a normal MCV.[15] The hemocytometer averages the 2 populations, macrocytic vitamin B12–deficient red blood cells and microcytic iron-deficient red blood cells, producing a normocytic value. Increased red cell distribution width (RDW) indicates the presence of different red blood cell populations.
Hypoproliferative Macrocytic Anemia (MCV > 100 fL)
Macrocytic anemias are subdivided into megaloblastic and nonmegaloblastic causes. Megaloblastic anemias arise from impaired DNA synthesis, producing characteristic oval macrocytes and hypersegmented neutrophils on peripheral smear (see Image. Macrocytic Anemia).[16]
Megaloblastic causes:
- Vitamin B12 (cobalamin) deficiency: Causes include pernicious anemia (autoimmune gastritis with intrinsic factor deficiency), dietary insufficiency (vegan or vegetarian diets), ileal resection or disease, and medications (metformin, proton pump inhibitors).[17]
- Folate deficiency: Causes include poor dietary intake, alcohol use disorder, medications (methotrexate, trimethoprim, phenytoin), increased demand (pregnancy, hemolytic anemias), and malabsorption.[15] Please see StatPearls' companion reference, "Folic Acid Deficiency," for further information.
Nonmegaloblastic causes:
- Myelodysplastic syndromes (MDS): These clonal hematopoietic stem cell disorders were reclassified by the 2022 World Health Organization 5th edition and the International Consensus Classification as myelodysplastic neoplasms, incorporating molecular features, including the splicing factor 3b subunit 1 gene (SF3B1) and the tumor protein p53 gene (TP53) mutations, into the diagnostic criteria.[18][19]
- Alcohol use disorder and alcoholic liver disease: Mechanisms include direct toxic effects on erythroid precursors, folate deficiency, and altered membrane lipid metabolism; macrocytosis may persist for weeks after abstinence due to the long erythrocyte lifespan.[20]
- Hypothyroidism: Thyroid hormones stimulate erythropoietin gene expression and directly enhance erythroid precursor proliferation; deficiency leads to decreased erythropoietic drive and may produce normocytic or macrocytic anemia.[21]
Drug-induced macrocytosis: Macrocytosis may occur with agents that interfere with DNA synthesis or erythroid maturation, including hydroxyurea, azathioprine, zidovudine, valproic acid, and fluoropyrimidines such as 5-fluorouracil. Drug-induced macrocytosis may be megaloblastic or nonmegaloblastic. Agents that interfere with DNA synthesis or folate metabolism, including methotrexate, trimethoprim, hydroxyurea, azathioprine, zidovudine, and fluoropyrimidines, may produce megaloblastic changes, whereas other medications, such as valproic acid and some antiretroviral agents, may cause macrocytosis through nonmegaloblastic or incompletely defined mechanisms.[16][22]
Please see StatPearls' companion references, "Vitamin B12 Deficiecy," "Folate Deficiency," and "Myelodysplastic Syndrome", for further information.
Hyperproliferative Anemia (Reticulocyte Production Index ≥ 3)
An elevated RPI indicates an appropriate bone marrow response to anemia, typically seen in hemolysis or acute blood loss. Hemolytic anemias are categorized by the site of RBC destruction and by intrinsic versus extrinsic mechanisms.[23]
Extravascular hemolysis (reticuloendothelial system mediated):
- Hemoglobinopathies: sickle cell disease, unstable hemoglobin variants [24]
- Membranopathies: hereditary spherocytosis, hereditary elliptocytosis; caused by defects in cytoskeletal or membrane proteins (spectrin, ankyrin, band 3) leading to loss of membrane surface area and spherocyte formation [23]
- Enzymopathies: glucose-6-phosphate dehydrogenase (G6PD) deficiency, pyruvate kinase deficiency [25]
- Autoimmune hemolytic anemia (AIHA): warm type (IgG-mediated, extravascular predominant) and cold agglutinin disease (IgM/complement-mediated); mixed-type AIHA with both warm and cold antibodies also occurs [26]
- Drug-induced hemolysis: hapten, immune complex, and autoantibody mechanisms; implicated drugs include cephalosporins, penicillins, nonsteroidal anti-inflammatory drugs, and methyldopa [26]
- Hypersplenism: sequestration and accelerated destruction of red cells in an enlarged spleen; common in portal hypertension and hematologic malignancies [23]
Intravascular hemolysis:
- Thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS): Thrombotic microangiopathies with microangiopathic hemolytic anemia and thrombocytopenia; TTP results from the a disintegrin and metalloproteinase with thrombospondin type 1 motif 13 (ADAMTS13) gene deficiency.[27]
- Disseminated intravascular coagulation (DIC): Systemic activation of coagulation with consumption of platelets and clotting factors; fibrin strand deposition causes mechanical fragmentation of erythrocytes.[23]
- Paroxysmal nocturnal hemoglobinuria (PNH): This acquired glycosylphosphatidylinositol anchor deficiency renders erythrocytes susceptible to complement-mediated lysis.[28]
- Mechanical hemolysis: Prosthetic heart valves, extracorporeal membrane oxygenation circuits, and march hemoglobinuria cause shear-stress fragmentation of erythrocytes, producing schistocytes on peripheral blood smear.[23]
- Severe infections: Malaria (direct parasitization and splenic clearance), Clostridium perfringens (phospholipase-mediated lysis), and babesiosis may produce both intravascular and extravascular hemolysis.[23]
- Transfusion reactions: Acute hemolytic transfusion reactions result from ABO-incompatible antibodies, causing complement-mediated intravascular hemolysis.[23]
- Toxins and venoms: Snake envenomation, copper toxicity (Wilson disease), and arsine gas exposure can cause direct oxidative damage to erythrocyte membranes.[23]
Acute blood loss: Acute hemorrhage from trauma, a surgical procedure, or gastrointestinal tract bleeding produces a hyperproliferative anemia because the bone marrow increases erythropoietic output in response to tissue hypoxia and elevated erythropoietin. Notably, hemoglobin may initially appear normal in acute hemorrhage before hemodilution develops over 24 to 72 h.[6]
Please see StatPearls' companion references, "Sickle Cell Disease," "Glucose-6-Phosphate Dehydrogenase Deficiency," "Laboratory Evaluation of Immune Hemolytic Anemias," "Thrombotic Thrombocytopenic Purpura," and "Paroxysmal Nocturnal Hemoglobinuria," for further information.
Epidemiology
According to the Global Burden of Disease Study 2021, anemia affected approximately 1.92 billion people worldwide, corresponding to a global prevalence of 24.3% across all ages.[7] Women are affected more frequently, with a prevalence of 31.2% compared with 17.5% in men; among women of reproductive age (15 to 49 years), the prevalence reaches 33.7%. Dietary iron deficiency accounts for approximately 66% of total anemia cases globally, comprising an estimated 444 million cases in men and 825 million in women. Children younger than 5 years are among the most affected, with iron deficiency being the leading cause, though hemoglobinopathies, malaria, and other infectious diseases are important contributors in endemic regions.[1]
In the United States, data from the National Health and Nutrition Examination Survey (NHANES) from 2003 to 2012 showed an overall prevalence of anemia of approximately 5.6%, with significant variation by age, sex, and race and ethnicity. Black individuals have a higher prevalence than White individuals, partly attributable to the higher frequency of hemoglobin variants and thalassemia traits in this population.[29] Anemia prevalence increases markedly with advancing age, affecting more than 10% of community-dwelling adults aged 65 years and older, more than 40% of hospitalized older adult patients, and up to 47% of nursing home residents.[30]
Among older adults with anemia, the etiology is divided roughly into thirds, a framework termed the rule of thirds: approximately one-third have nutritional deficiency anemia (iron, folate, or vitamin B12), one-third have chronic kidney disease or chronic inflammation, and one-third have anemia that remains unexplained even after thorough evaluation.[31] Unexplained anemia of aging may reflect age-related clonal hematopoiesis, subclinical inflammation (inflammaging), declining stem cell reserve, or relative androgen deficiency.[30][32]
Pregnancy represents a high-risk state for anemia, affecting approximately 32 million pregnant women globally and contributing to an estimated 115,000 maternal deaths and 591,000 perinatal deaths annually. The global prevalence of anemia in pregnancy remains more than twice the World Health Organization target of 15%.[33] Iron deficiency accounts for the majority of cases, exacerbated by the physiologic increase in plasma volume that exceeds the expansion of red cell mass during gestation.
Pathophysiology
Anemia results from a fundamental imbalance between red blood cell production and red blood cell loss or destruction. Red blood cells have a typical lifespan of approximately 120 days, and the bone marrow normally replaces roughly 0.8% to 1.0% of circulating red blood cells daily. The 3 principal pathophysiologic mechanisms are described below.[6]
Decreased or Ineffective Erythropoiesis
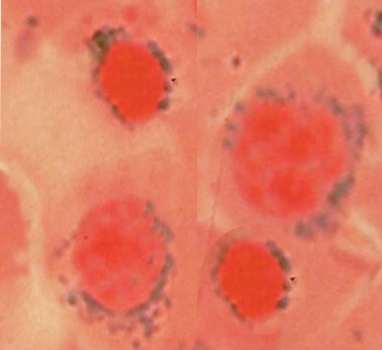
Nutritional deficiencies deprive erythroid precursors of essential substrates. Iron deficiency leads to reduced hemoglobin synthesis and microcytic, hypochromic erythrocytes (see Image. Hypochromic Microcytic Anemia). Iron is required not only for heme synthesis but also for DNA synthesis enzymes (eg, ribonucleotide reductase), contributing to impaired erythroid proliferation.[34] Vitamin B12 and folate are essential cofactors for thymidylate synthase and methionine synthase, both of which are critical for DNA synthesis. These deficiencies lead to nuclear-cytoplasmic dyssynchrony, in which the nucleus matures slowly while the cytoplasm develops normally, resulting in large erythroblasts and intramedullary apoptosis, a hallmark of megaloblastic anemia.[17]
In anemia of inflammation or anemia of chronic disease, the hepatic peptide hormone hepcidin plays a central role. Inflammatory cytokines, particularly interleukin-6 (IL-6), upregulate hepcidin production via the Janus kinase–signal transducer and activator of transcription 3 (JAK-STAT3) signaling pathway. Hepcidin binds to and degrades ferroportin, the sole known cellular iron exporter, on enterocytes, macrophages, and hepatocytes. This process results in decreased intestinal iron absorption and iron sequestration by the reticuloendothelial system, leading to functional iron deficiency despite adequate or elevated total-body iron stores.[3] Additional mechanisms in anemia of inflammation include direct cytokine-mediated suppression of erythroid progenitors (tumor necrosis factor-α and interferon-γ), relative EPO deficiency, and shortened RBC lifespan.[35]
In CKD, inadequate EPO production by diseased kidneys leads to insufficient stimulation of erythroid progenitors. Uremic toxins further impair erythropoiesis and shorten RBC survival.[11] In bone marrow failure syndromes (eg, aplastic anemia), immune-mediated destruction of hematopoietic stem cells results in pancytopenia.[12] In myelodysplastic syndromes and thalassemia major, erythropoiesis is active but ineffective, with erythroid precursors produced in abundance yet undergoing premature intramedullary apoptosis.[19]
Increased Red Blood Cell Destruction (Hemolysis)
Hemolytic anemias are characterized by premature red blood cell destruction, either within the reticuloendothelial system (extravascular) or within the vasculature (intravascular). Intrinsic (corpuscular) causes include inherited defects of the red blood cell membrane (hereditary spherocytosis, elliptocytosis), hemoglobin structure (sickle cell disease, unstable hemoglobins), or enzyme systems (glucose-6-phosphate dehydrogenase deficiency, pyruvate kinase deficiency). Extrinsic (extracorpuscular) causes include immune-mediated destruction (autoantibodies, alloantibodies), mechanical shear stress (microangiopathic hemolytic anemias, prosthetic valves), direct toxic or infectious injury (malaria, Clostridium perfringens infection), and complement-mediated lysis (paroxysmal nocturnal hemoglobinuria.[23] Compensatory bone marrow hyperplasia can increase erythropoiesis by 6- to 8-fold; anemia develops only when the rate of destruction exceeds this maximal compensatory capacity. The bone marrow response is reflected by reticulocytosis, which typically becomes apparent 3 to 5 days after the onset of hemolysis.
Blood Loss
Acute hemorrhage (trauma, surgical procedures, gastrointestinal tract bleeding, or ruptured ectopic pregnancy) leads to rapid intravascular volume depletion. Anemia becomes apparent on laboratory testing only after transcapillary refill and hemodilution occur over 24 to 72 h. Chronic occult blood loss, most commonly from the gastrointestinal tract (peptic ulcer disease, colorectal neoplasia, angiodysplasia, or hookworm infestation) or the genitourinary tract (menorrhagia, renal cell carcinoma, or schistosomiasis), progressively depletes iron stores, eventually producing iron deficiency anemia.[36] Gastrointestinal evaluation is recommended for all men and postmenopausal women with iron-deficiency anemia and should be considered for premenopausal women when no clear gynecologic source is identified.[37]
History and Physical
Symptom onset correlates with the rapidity and severity of hemoglobin decline. Chronic, slowly progressive anemia may be remarkably well-tolerated due to compensatory cardiovascular and respiratory adaptations.
- Fatigue, weakness, exercise intolerance, and dyspnea on exertion are the most common presenting concerns.[6]
- Palpitations, lightheadedness, presyncope, or syncope reflect decreased oxygen delivery and compensatory tachycardia.[6]
- Chest pain and exertional angina occur particularly in patients with underlying coronary artery disease; they may precipitate acute coronary syndrome when hemoglobin is less than 7 to 8 g/dL.[38]
- Restless legs syndrome is strongly associated with iron deficiency even in the absence of frank anemia; symptoms typically respond to iron repletion.[39]
- Cognitive impairment and difficulty concentrating are particularly common in older adult and pediatric populations; iron deficiency impairs neurotransmitter synthesis.[30]
- Pica and pagophagia are relatively specific to iron deficiency; their pathophysiology is poorly understood, but they resolve rapidly with iron supplementation.[40]
- Asymptomatic anemia may occur because mild anemia may be entirely asymptomatic and discovered incidentally on routine complete blood count testing; most patients become symptomatic when hemoglobin falls below 7.0 g/dL.[6]
Physical Examination Findings
- General: Findings include pallor of conjunctivae, oral mucosa, and palmar creases; conjunctival pallor has a positive likelihood ratio of 4.5 for significant anemia (hemoglobin ≤ 9 g/dL); pallor of palmar creases has high specificity.[41]
- Head, Eyes, Ears, Nose, and Throat: Findings include glossitis and angular cheilitis (iron, vitamin B12, or folate deficiency), scleral icterus (hemolysis), lymphadenopathy (hematologic malignant neoplasm), and boxcar or sausage-shaped retinal veins (hyperviscosity in myeloproliferative disease).[17][40]
- Cardiovascular: Findings include resting tachycardia, wide pulse pressure, systolic flow murmur, and signs of high-output heart failure in severe chronic anemia.[38]
- Abdominal: Findings include splenomegaly (hemolytic anemia, myeloproliferative disease, or portal hypertension), hepatomegaly (liver disease, myelofibrosis, or extramedullary hematopoiesis), and surgical scars (gastrectomy scar raises concern for vitamin B12 deficiency; cholecystectomy scar may suggest chronic hemolysis).[6]
- Neurologic: Findings include impaired proprioception, decreased vibratory sensation, a positive Romberg test, an ataxic gait (subacute combined degeneration due to vitamin B12 deficiency), and peripheral neuropathy.[17]
- Skin and nails: Findings include koilonychia (spooning of nails, suggesting iron deficiency), petechiae and purpura (thrombocytopenia or disseminated intravascular coagulation), dermatitis herpetiformis (celiac disease), and leg ulcers (sickle cell disease.[24][40]
- Rectal and pelvic: Evaluation should assess for hemorrhoids, rectal masses, and heavy vaginal bleeding as sources of blood loss.[42]
Evaluation
A systematic, stepwise approach to the anemic patient reduces unnecessary testing and accelerates diagnosis. The following 4-step framework is recommended:
Step 1: Confirm Anemia and Review the Complete Blood Count
The CBC with differential provides the hemoglobin, hematocrit, RBC count, MCV, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, red cell distribution width (RDW), white blood cell count with differential, and platelet count. The RDW quantifies anisocytosis (variation in red cell size); it is characteristically elevated in iron deficiency anemia and often normal in thalassemia trait, providing a useful (though not absolute) discriminator.[43] Abnormalities in more than one cell line (eg, bicytopenia or pancytopenia) should raise concern for bone marrow pathology, including aplastic anemia, MDS, leukemia, or marrow infiltration.
Step 2: Calculate the Corrected Reticulocyte Count
The reticulocyte production index (RPI) corrects the reticulocyte percentage for the degree of anemia:
Corrected Reticulocyte Count (%) = Reticulocyte Count (%) × (Patient Hematocrit / Normal Hematocrit)
Reticulocyte Production Index = Corrected Reticulocyte Count / Maturation Factor
(1.0 for hematocrit 45%, 1.5 for 35%, 2.0 for 25%, and 2.5 for 15%)
Use 45% as normal hematocrit for men and 40% for women. An RPI of 2 or greater indicates an appropriate marrow response and suggests hemolysis or acute blood loss. An RPI less than 2 indicates a hypoproliferative marrow response that fails to compensate adequately for the anemia. Please see StatPearls' companion reference, "Histology, Reticulocytes," for further information.
Step 3: Classify by MCV and Obtain Targeted Studies for Microcytic Anemia (MCV < 80 fL)
Iron studies: Serum iron, ferritin, total iron-binding capacity, and transferrin saturation form the core iron panel. Serum ferritin is the most useful single test for iron deficiency: a ferritin less than 30 ng/mL has high sensitivity in the general population, while the American Gastroenterological Association recommends a cutoff of less than 45 ng/mL for improved diagnostic sensitivity in the gastrointestinal tract evaluation.[37][44] In inflammatory states, ferritin loses specificity because ferritin is an acute-phase reactant; a ferritin less than 100 ng/mL with transferrin saturation less than 20% is suggestive of concomitant true iron deficiency in the setting of inflammation.[45] The American Society of Hematology 2025 draft guidelines recommend using both ferritin and transferrin saturation, rather than ferritin alone, to evaluate iron deficiency in adults with anemia of inflammation.
The soluble transferrin receptor is elevated in iron deficiency but remains within the reference range in anemia of inflammation or anemia of chronic disease, making soluble transferrin receptor useful when conventional iron studies are equivocal. The soluble transferrin receptor and log ferritin index provides an integrated measure of body iron stores that is less affected by inflammation.[43] Reticulocyte hemoglobin content (CHr or Ret-He), available on many modern analyzers, reflects the iron available for recent erythropoiesis and can detect functional iron deficiency earlier than conventional iron parameters.
Hemoglobin electrophoresis is indicated when thalassemia is suspected. Elevated hemoglobin A2 (greater than 3.5%) or hemoglobin F is characteristic of β-thalassemia trait, while α-thalassemia trait shows a normal electrophoresis pattern result; molecular testing confirms the diagnosis.[8] Serum lead levels should be obtained when occupational or environmental exposure is suspected; basophilic stippling on smear is a classic (though not sensitive) finding.[10]
Normocytic anemia (MCV 80–100 fL):
- Comprehensive metabolic panel: blood urea nitrogen, creatinine for chronic kidney disease assessment, liver function tests, and albumin
- Inflammatory markers: C-reactive protein, erythrocyte sedimentation rate, and ferritin, which may be elevated as an acute-phase reactant in anemia of inflammation
- Serum protein electrophoresis with immunofixation and serum free light chains if multiple myeloma or another plasma cell dyscrasia is suspected
- Erythropoietin level is helpful to distinguish chronic kidney disease–associated anemia (low erythropoietin levels) from other causes
- Bone marrow biopsy is indicated for suspected aplastic anemia, myelofibrosis, myelophthisic processes, or unexplained cytopenias
- Peripheral smear: A leukoerythroblastic picture (nucleated red blood cells and immature granulocytes) suggests marrow infiltration [6]
Macrocytic anemia (MCV > 100 fL):
Serum vitamin B12 and folate levels should be obtained first. Methylmalonic acid and homocysteine help distinguish between deficiencies: both are elevated in vitamin B12 deficiency, whereas only homocysteine rises in folate deficiency (methylmalonic acid remains within the reference range). Methylmalonic acid is particularly useful when the vitamin B12 level is in the indeterminate range (200 to 400 pg/mL).[17]
- Thyroid function tests: thyroid-stimulating hormone and free thyroxine for hypothyroidism
- Hepatic function panel: γ-glutamyl transferase, and assessment of alcohol use
- Peripheral smear: Hypersegmented neutrophils (5 or more lobes, or any with 6 or more lobes) and oval macrocytes suggest megaloblastic anemia
- Bone marrow biopsy if myelodysplastic syndromes are suspected (cytopenias, dysplastic morphology, or blasts on smear).[18] The presence of bilinear cytopenia, defined as decreases in 2 of 3 cell lines, including platelets, white blood cells, or red blood cells, on the peripheral blood smear should strongly suggest myelodysplasia.
Step 4: Evaluate for Hemolysis (if RPI ≥ 2)
Confirm hemolysis with the hemolytic markers panel: elevated lactate dehydrogenase, elevated indirect or unconjugated bilirubin, decreased or undetectable haptoglobin, and elevated reticulocyte count. Haptoglobin is the most sensitive marker because free hemoglobin binding consumes haptoglobin.[23]
Differentiate intravascular from extravascular hemolysis:
- Extravascular: spherocytes on smear, negative results for urine hemoglobin and hemosiderin
- Intravascular: elevated plasma-free hemoglobin, positive results for urine hemoglobin, urine hemosiderin, schistocytes (in microangiopathic causes), and hemoglobinemia
Peripheral blood smear morphology provides critical etiologic clues:
- Spherocytes suggest autoimmune hemolytic anemia (direct antiglobulin test [DAT]–positive) or hereditary spherocytosis (DAT–negative).
- Schistocytes (fragmented cells) suggest thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, disseminated intravascular coagulation, mechanical hemolysis, malignant hypertension, or hemolysis, elevated liver enzymes, and low platelet count syndrome.
- Bite cells or blister cells suggest glucose-6-phosphate dehydrogenase deficiency due to oxidant injury.
- Target cells suggest hemoglobinopathies, thalassemia, liver disease, or postsplenectomy status.
- Sickle cells (drepanocytes) suggest sickle cell disease.
- Acanthocytes (spur cells) suggest severe liver disease or abetalipoproteinemia.
- Parasitic inclusions suggest malaria, babesiosis, or bartonellosis.[6][23]
The direct antiglobulin test (DAT, or direct Coombs test) is essential when spherocytes are present. A positive DAT result confirms immune-mediated hemolysis (autoimmune hemolytic anemia). A negative direct antiglobulin test result in the setting of spherocytes suggests hereditary spherocytosis or, less commonly, direct antiglobulin test–negative autoimmune hemolytic anemia, which occurs in approximately 5% to 10% of autoimmune hemolytic anemia cases and may require more sensitive testing techniques (eg, flow cytometry, mitogen-stimulated direct antiglobulin testing).[23] High-sensitivity flow cytometry for glycosylphosphatidylinositol-anchored proteins (CD55, CD59) should be performed to evaluate for paroxysmal nocturnal hemoglobinuria in patients with unexplained hemolysis, particularly when accompanied by thrombosis or cytopenias.[28]
Additional Investigations
The American Gastroenterological Association 2020 Clinical Practice Guidelines recommend bidirectional endoscopy (esophagogastroduodenoscopy and colonoscopy) in all asymptomatic postmenopausal women and men with iron deficiency anemia, and suggest it in premenopausal women with iron deficiency anemia.[37] The American Gastroenterological Association 2024 Clinical Practice Update further recommends that intravenous iron be used preferentially in patients with inflammatory bowel disease, postbariatric surgical procedures, or intolerable oral iron adverse effects, and that gastrointestinal tract evaluation should not be deferred until after iron repletion.[46] Additional evaluation may include testing for Helicobacter pylori infection, which is associated with iron-deficiency anemia even without peptic ulcer; celiac disease serologies (tissue transglutaminase IgA with total IgA); pelvic ultrasonography for evaluation of menorrhagia; and cross-sectional imaging when a malignant neoplasm or an internal hemorrhage is suspected.[37]
Treatment / Management
Management of anemia is directed at the underlying cause. The following section provides an overview of general management principles and key evidence-based recommendations; detailed treatment protocols are discussed in the corresponding subtype-specific articles.
Transfusion Thresholds
Multiple landmark randomized controlled trials established the safety and efficacy of restrictive transfusion strategies in most clinical settings:
- Transfusion Requirements in Critical Care (TRICC) trial (1999): In this trial, 838 critically ill patients in the intensive care unit were randomized to restrictive (hemoglobin trigger ≤ 7 g/dL) or liberal (hemoglobin trigger ≤ 10 g/dL) strategies; the restrictive strategy was noninferior and potentially superior in younger, less acutely ill patients.[47]
- Transfusion Requirements in Septic Shock (TRISS) trial (2014): In this trial, 998 patients with septic shock were assigned to a restrictive threshold (hemoglobin ≤ 7 g/dL) or liberal threshold (hemoglobin ≤ 9 g/dL); 90-day mortality did not differ, with 50% fewer transfusions in the restrictive group.[48]
- Functional Outcomes in Cardiovascular Patients Undergoing Surgical Hip Fracture Repair (FOCUS) trial (2011): In this trial, 2016 high-risk patients after hip surgical procedures with cardiovascular disease or risk factors were assigned to a liberal threshold (hemoglobin ≥ 10 g/dL) or restrictive threshold (hemoglobin ≤ 8 g/dL or symptomatic); functional recovery and 60-day mortality did not differ.[49] (A1)
Current consensus recommendations: Clinicians should maintain hemoglobin levels greater than 7 g/dL in hemodynamically stable patients without cardiac disease. Patients with acute coronary syndrome or significant cardiovascular disease may benefit from a higher threshold of hemoglobin greater than 8 g/dL, based on subgroup analyses suggesting increased ischemic events with restrictive strategies in this population.[50] Patient blood management programs emphasize a multimodal approach, including preoperative anemia optimization, intraoperative blood conservation, and evidence-based transfusion triggers.(A1)
Iron Deficiency Anemia
Oral iron remains the first-line therapy for most patients with iron deficiency. Ferrous sulfate (325-mg tablet containing 65 mg of elemental iron) is the most commonly prescribed formulation. Results from landmark studies by Moretti et al in 2015 demonstrated that oral iron doses of 60 mg or more elemental iron increase serum hepcidin for approximately 24 h, reducing fractional iron absorption from subsequent doses by 35% to 45%.[51] Results from a study by Stoffel et al in 2017 confirmed that alternate-day dosing provides superior fractional iron absorption compared with consecutive-day or twice-daily dosing.[52] Based on this evidence, a single morning dose on alternate days is now recommended to maximize absorption and minimize adverse gastrointestinal effects (nausea, constipation, a metallic taste, and black stools). Expected response includes a rise in reticulocyte count within 7 to 10 days and normalization of hemoglobin within 6 to 8 weeks. Iron supplementation should continue for at least 3 months after hemoglobin normalization to fully replenish iron stores (target ferritin > 50 to 100 ng/mL).[53](A1)
Intravenous iron is indicated in patients who are intolerant of oral iron, have malabsorptive conditions (celiac disease, inflammatory bowel disease, or postbariatric surgical procedures), require rapid iron repletion, or have ongoing blood loss exceeding oral replacement capacity. The American Gastroenterological Association 2024 update recommends intravenous formulations capable of total dose repletion in 1 or 2 sessions.[46] Contemporary formulations include:
- Ferric carboxymaltose: This formulation may be given at up to 750 mg per infusion (15 mg/kg), with 2 doses separated by at least 7 days. Ferric carboxymaltose has a known risk of transient hypophosphatemia (up to 50% of patients) via increased fibroblast growth factor 23, which usually resolves within weeks but can be clinically significant with repeated dosing.
- Ferric derisomaltose: This formulation may be given at up to 1000 to 1500 mg in a single infusion. Ferric derisomaltose has efficacy equivalent to that of ferric carboxymaltose without the risk of hypophosphatemia and an excellent safety profile.
- Iron sucrose: This formulation is given at 200 mg per infusion, requiring multiple sessions for full repletion. Clinicians commonly use iron sucrose in patients with chronic kidney disease.[11] (A1)
Please see StatPearls' companion reference, "Iron Deficiency Anemia," for further information.
Vitamin B12 and Folate Deficiency
Vitamin B12 deficiency is treated with intramuscular cyanocobalamin (1000 µg daily for 7 days, then weekly for 4 weeks, then monthly maintenance) or, for dietary deficiency, high-dose oral vitamin B12 (1000 to 2000 µg/d), which achieves comparable efficacy via passive diffusion (approximately 1% of the oral dose is absorbed independent of intrinsic factor).[17] Folate deficiency is corrected with oral folic acid 1 to 5 mg/d. Clinicians must exclude vitamin B12 deficiency before initiating folate replacement, because folate may partially correct the hematologic abnormalities of vitamin B12 deficiency while allowing progression of irreversible neurologic damage (subacute combined degeneration of the spinal cord).[15]
Please see StatPearls' companion reference, "Vitamin B12 Deficiency," and "Folate Deficiency," for further information.
Anemia of Inflammation or Chronic Disease
Management centers on treating the underlying inflammatory, infectious, or neoplastic condition. For patients with concurrent true iron deficiency (ferritin < 100 ng/mL and transferrin saturation < 20%), intravenous iron may be beneficial; oral iron is typically ineffective due to hepcidin-mediated blockade of intestinal absorption.[15] Erythropoiesis-stimulating agents (ESAs), including epoetin alfa and darbepoetin alfa, are used in selected patients with chronic kidney disease and cancer-related anemia, with careful attention to thromboembolic risk and target hemoglobin ranges. The Kidney Disease: Improving Global Outcomes (KDIGO) 2026 guidelines recommend ESAs as first-line pharmacotherapy for chronic kidney disease–associated anemia, rather than hypoxia-inducible factor prolyl hydroxylase inhibitors, once correctable causes have been addressed, with treatment initiation suggested when hemoglobin falls to 9.0 to 10.0 g/dL in dialysis-dependent patients.[11](A1)
Hypoxia-inducible factor prolyl hydroxylase inhibitors (HIF-PHIs) such as roxadustat, daprodustat, and vadadustat represent a newer class of oral agents that stimulate endogenous erythropoietin production and improve iron utilization. Results from multiple phase 3 trials demonstrated noninferiority to ESAs for hemoglobin correction and maintenance. Kidney Disease: Improving Global Outcomes 2026 recommends ESAs over HIF-PHIs as first-line therapy, based on more extensive long-term safety data, but HIF-PHIs may be preferred in selected situations (eg, ESA hyporesponsiveness, patient preference for oral therapy).[11](A1)
Please see StatPearls' companion reference, "Normochromic Normocytic Anemia" and "Anemia of Chronic Kidney Disease," for further information.
Hemolytic Anemias
Management depends on the underlying mechanism:
Warm autoimmune hemolytic anemia: Corticosteroids (prednisone 1 to 2 mg/kg/d) are first-line therapy, achieving an initial response in 70% to 85% of patients. Rituximab (anti-CD20) is recommended as second-line therapy. Splenectomy is reserved for refractory disease. Mycophenolate mofetil, azathioprine, and fostamatinib are additional options.[54]
Cold agglutinin disease: Treatment includes avoidance of cold exposure and rituximab-based therapy for symptomatic patients; corticosteroids and splenectomy are generally ineffective.[54]
Glucose-6-phosphate dehydrogenase deficiency: Treatment includes avoidance of oxidative triggers, such as fava beans and certain medications (eg, dapsone, primaquine, rasburicase, and sulfonamides); supportive care; and transfusion during acute hemolytic episodes.[25]
Hereditary spherocytosis: Treatment includes folic acid supplementation, splenectomy (preferably partial in children) for moderate to severe disease, and cholecystectomy for symptomatic pigmented gallstones.
Thrombotic thrombocytopenic purpura: Urgent therapeutic plasma exchange is the cornerstone of treatment. Caplacizumab, an anti–von Willebrand factor nanobody, significantly reduces time to platelet recovery, recurrences, and thrombotic thrombocytopenic purpura–related death when added to therapeutic plasma exchange and immunosuppression (Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura [HERCULES] trial).[27]
Paroxysmal nocturnal hemoglobinuria: Complement inhibitors, including eculizumab (anti-C5) and ravulizumab (long-acting anti-C5), are first-line therapies that dramatically reduce intravascular hemolysis and transfusion requirements.[23][28]
Please see StatPearls' companion references, "Glucose-6-Phosphate Dehydrogenase Deficiency," "Laboratory Evaluation of Immune Hemolytic Anemias," "Thrombotic Thrombocytopenic Purpura," and "Paroxysmal Nocturnal Hemoglobinuria," for further information.
Sickle Cell Disease
Hydroxyurea remains the cornerstone disease-modifying therapy, increasing fetal hemoglobin production and reducing vaso-occlusive crises, acute chest syndrome episodes, and transfusion requirements.[24] L-glutamine is US Food and Drug Administration (FDA)–approved as an adjunctive therapy to reduce acute complications. Notably, voxelotor (a hemoglobin S polymerization inhibitor) was voluntarily withdrawn from the market in 2023 due to safety concerns identified in postmarketing studies, and crizanlizumab (an anti–P-selectin antibody) was withdrawn in 2024 after confirmatory trials failed to demonstrate efficacy.
Gene therapy represents a transformative advance: in December 2023, the FDA approved 2 gene therapy products for patients aged 12 years or older with recurrent vaso-occlusive events, exagamglogene autotemcel (Casgevy, the first clustered regularly interspaced short palindromic repeats–CRISPR-associated protein 9 [CRISPR-Cas9]–based therapy, targeting the B-cell lymphoma/leukemia 11A (BCL11A) gene to upregulate fetal hemoglobin) and lovotibeglogene autotemcel (Lyfgenia, a lentiviral vector–based therapy introducing an antisickling β-globin). Both achieved vaso-occlusive event–free outcomes in approximately 94% of trial participants. Allogeneic hematopoietic stem cell transplant from a matched sibling donor remains a curative option for eligible patients.[55][56](B3)
Please see StatPearls' companion reference, "Sickle Cell Disease," for further information.
Thalassemia Syndromes
Management depends on disease severity. Transfusion-dependent thalassemia (previously thalassemia major) requires regular transfusion programs with iron chelation therapy (deferasirox, deferoxamine, or deferiprone) to prevent iron overload–related organ damage. Luspatercept, a first-in-class erythroid maturation agent, is approved for patients with transfusion-dependent thalassemia who require regular transfusions, reducing transfusion burden by approximately 33%. Non–transfusion-dependent thalassemia is treated with surveillance, folic acid supplementation, and iron chelation when indicated. Allogeneic hematopoietic stem cell transplant remains the only established curative approach, though gene therapy trials have shown promise.[57]
Please see StatPearls' companion reference, "Thalassemia," for further information.
Bone Marrow Failure Syndromes
Aplastic anemia: The British Society for Haematology 2024 guidelines recommend horse antithymocyte globulin plus cyclosporine as first-line immunosuppressive therapy, with the addition of eltrombopag, a thrombopoietin receptor agonist that has improved overall response rates. Allogeneic hematopoietic stem cell transplant from a matched sibling donor is preferred for younger patients (40 years or younger) with severe aplastic anemia.[12]
Myelodysplastic syndromes: Treatment is risk-stratified using the Revised International Prognostic Scoring System or the Molecular International Prognostic Scoring System. Lower-risk myelodysplastic syndromes are treated with supportive care (transfusions, erythropoietin), lenalidomide for del(5q) myelodysplastic syndromes, and luspatercept for ring sideroblast–positive subtypes. Higher-risk myelodysplastic syndromes are treated with hypomethylating agents (azacitidine, decitabine) and allogeneic hematopoietic stem cell transplant in transplant-eligible patients.[18][19] Please see StatPearls' companion references, "Aplastic Anemia," and "Myelodysplastic Syndrome, for further information.(B3)
Differential Diagnosis
Several conditions can mimic or confound the laboratory diagnosis of anemia:
- Hemodilution: Aggressive intravenous fluid resuscitation, pregnancy (physiologic hemodilution peaks at 28–34 weeks), massive splenomegaly, and paraproteinemia (altered Colter counter measurements) may cause a factitious reduction in hemoglobin concentration.[58][59][60]
- Specimen hemolysis during phlebotomy can produce artifacts in the complete blood count, including falsely elevated potassium and lactate dehydrogenase levels.[61]
- Acute hemorrhage: Hemoglobin may initially remain normal before fluid redistribution and transcapillary refill produce hemodilution over 24–72 hours.[62]
- Analytical interference: Lipemia, extreme leukocytosis, or cryoglobulins can affect automated CBC results.[63]
Prognosis
Prognosis is fundamentally linked to the underlying etiology and the timeliness of intervention:
Nutritional deficiency anemias: Nutritional deficiency anemias generally carry an excellent prognosis when identified and treated promptly. Iron supplementation should continue for at least 3 months after hemoglobin normalization to ensure repletion of iron stores. Vitamin B12 deficiency–related neurologic damage may be irreversible if treatment is delayed, underscoring the importance of early diagnosis.[64][65]
Anemia in older adults: Anemia in older adults is independently associated with increased all-cause mortality, hospitalization, falls, cognitive decline, functional impairment, and reduced quality of life, even at hemoglobin levels traditionally considered mild.[31]
Anemia of chronic kidney disease: Anemia of chronic kidney disease treated with erythropoiesis-stimulating agents and iron supplementation improves symptoms and quality of life, though overcorrection, defined as targeting hemoglobin greater than 13 g/dL, is associated with adverse cardiovascular outcomes, including stroke, heart failure, and death (per the Correction of Hemoglobin and Outcomes in Renal Insufficiency [CHOIR], Cardiovascular Risk Reduction by Early Anemia [CREATE], and treial to reduce cardiovascular events with Aranesp Therapy [TREAT] trials).[11]
Hemolytic anemias: Hemolytic anemias have variable prognoses, ranging from self-limited episodes, such as glucose-6-phosphate dehydrogenase deficiency, to life-threatening emergencies, such as thrombotic thrombocytopenic purpura, requiring immediate intervention. Mortality in thrombotic thrombocytopenic purpura has decreased from greater than 90% to 20% or less with the advent of therapeutic plasma exchange and caplacizumab.[66]
Sickle cell disease: Sickle cell disease prognosis has improved dramatically with hydroxyurea, comprehensive care, and gene therapy, but median life expectancy remains reduced compared with the general population.[67]
Complications
Unrecognized or inadequately treated anemia can lead to significant morbidity across multiple organ systems:
Cardiovascular: Complications include compensatory tachycardia, systolic flow murmurs, high-output heart failure, exacerbation of angina, myocardial infarction, arrhythmias, and left ventricular hypertrophy in chronic anemia. Heart failure is the leading cause of morbidity in patients with severe thalassemia and chronic transfusion-dependent anemia.[11][68]
Obstetric: Maternal anemia increases the risk of preterm birth (relative risk, 1.21), low birth weight (relative risk, 1.31), cesarean delivery (relative risk, 1.28), postpartum hemorrhage, and maternal mortality. Neonatal anemia and impaired iron endowment may also result.[33][69]
Neurologic: Chronic iron deficiency in infancy and childhood is associated with cognitive impairment and developmental delays that may not be fully reversible with treatment. Vitamin B12 deficiency can cause irreversible subacute combined degeneration of the spinal cord, with demyelination of the posterior columns and lateral corticospinal tracts.[70][71]
Perioperative: Preoperative anemia is an independent risk factor for perioperative morbidity, increased transfusion requirements, prolonged hospital stay, and mortality across surgical specialties.[72]
Other: Complications include restless legs syndrome and Plummer-Vinson syndrome, characterized by iron deficiency, esophageal webs, and dysphagia. Chronically transfused patients may develop iron overload complications, including cardiac siderosis, hepatic fibrosis, and endocrinopathies.[73][74][75][74]
Consultations
Subspecialty referral should be considered in the following scenarios:
Hematology: Referral is indicated for suspected hemolytic anemia requiring specialized evaluation; bone marrow failure syndromes (aplastic anemia or myelodysplastic syndromes); unexplained anemia persisting after routine evaluation; need for bone marrow biopsy; hemoglobinopathy treatment; or thrombotic thrombocytopenic purpura and hemolytic uremic syndrome requiring therapeutic plasma exchange.
Gastroenterology: Referral is indicated for iron-deficiency anemia without a clear source; bidirectional endoscopy per American Gastroenterological Association guidelines; suspected celiac disease; inflammatory bowel disease; or obscure gastrointestinal tract bleeding requiring capsule endoscopy or deep enteroscopy.
Nephrology: Referral is indicated for anemia of chronic kidney disease requiring erythropoiesis-stimulating agent therapy, intravenous iron treatment, or consideration of hypoxia-inducible factor prolyl hydroxylase inhibitor therapy per Kidney Disease: Improving Global Outcomes 2026 guidelines.
Gynecology: Referral is indicated for heavy menstrual bleeding refractory to medical treatment or evaluation for structural uterine pathology, including fibroids, polyps, or adenomyosis.
Oncology: Referral is indicated for anemia in the setting of known or suspected cancer or cancer-related anemia requiring erythropoiesis-stimulating agent therapy.
Deterrence and Patient Education
Effective patient education is integral to anemia management and prevention:
Iron deficiency: Patients with iron deficiency should be counseled on dietary sources of heme iron, including red meat, poultry, and fish, which have higher bioavailability, and nonheme iron, including legumes, fortified cereals, dark leafy greens, and tofu. Coadministration of vitamin C (ascorbic acid) enhances nonheme iron absorption by reducing ferric iron to ferrous iron in the gastrointestinal tract.
Iron absorption: Patients should be informed that tea, coffee, calcium supplements, and dairy products can decrease iron absorption and should be taken separately from iron supplements, ideally at least 2 h apart.
Oral iron therapy: Patients starting oral iron therapy should be educated on expected adverse effects, including metallic taste, constipation, nausea, and black stools, as well as the rationale for alternate-day dosing, which improves tolerability while maximizing absorption.
Intravenous iron candidacy: Patients should be advised to contact their clinician if they experience severe intolerance to oral iron, because they may be candidates for intravenous iron supplementation.
Vegetarian or vegan diets and bariatric surgical procedures: Patients following vegetarian or vegan diets are at risk of vitamin B12 deficiency and should be advised to consume vitamin B12- fortified foods or to supplement. Patients who have undergone bariatric surgical procedures, particularly Roux-en-Y gastric bypass, require lifelong monitoring and supplementation with iron, vitamin B12, and folate due to loss of absorptive surface area.
Hemoglobinopathies: Patients with hemoglobinopathies and their families benefit from genetic counseling and education regarding disease inheritance patterns, available screening, and reproductive options.
Glucose-6-phosphate dehydrogenase deficiency: Patients with glucose-6-phosphate dehydrogenase deficiency should receive a list of medications and foods to avoid and carry this information with them.
Pearls and Other Issues
Always review the peripheral blood smear when the etiology of anemia is not apparent from the initial laboratory evaluation; morphologic clues often direct the evaluation more efficiently than additional serologic testing.
Red cell distribution width: Red cell distribution width can help differentiate iron deficiency, which elevates it reflecting anisocytosis, from thalassemia trait, which often keeps it within the reference range reflecting uniform microcytosis. However, this distinction is not absolute and should not be used in isolation.
Mixed anemia: Concurrent iron and vitamin B12 deficiency may produce a deceptively normal mean corpuscular volume. Elevated red cell distribution width with a dimorphic red cell population on peripheral smear should raise suspicion for dual deficiency.
Anemia of inflammation: Ferritin is an acute-phase reactant and can be falsely within the reference range or elevated. Transferrin saturation less than 20%, in conjunction with ferritin 100 to 300 ng/mL, suggests coexisting true iron deficiency. The American Society of Hematology 2025 draft guidelines recommend using both ferritin and transferrin saturation together for this assessment.
Rectal and pelvic examination: Clinicians should not overlook rectal and pelvic examinations during the evaluation of anemia, as these examinations are frequently omitted in clinical practice yet can reveal important bleeding sources.
Celiac disease screening: Clinicians should screen for celiac disease with tissue transglutaminase immunoglobulin A and total immunoglobulin A in patients with unexplained iron deficiency, particularly when iron deficiency is refractory to oral iron supplementation or occurs in younger patients without obvious bleeding sources.
Reticulocyte hemoglobin content: Reticulocyte hemoglobin content is emerging as a useful early marker of functional iron deficiency, particularly in patients with chronic kidney disease receiving erythropoiesis-stimulating agent therapy, and is available on most modern automated hematology analyzers.
Oral iron dosing: Clinicians should consider alternate-day dosing rather than daily or twice-daily dosing to improve absorption and reduce gastrointestinal adverse effects, given evidence that oral iron induces hepcidin elevation lasting approximately 24 h.
Enhancing Healthcare Team Outcomes
Anemia management requires coordinated interprofessional collaboration to optimize patient outcomes. Primary care clinicians serve as the initial point of evaluation and triage, while hematologists, gastroenterologists, nephrologists, gynecologists, and oncologists contribute subspecialty expertise as warranted by the underlying etiology. Pharmacists play a vital role in patient counseling on iron and vitamin supplementation, medication interactions (eg, iron with levothyroxine, tetracyclines, or proton pump inhibitors), and adherence monitoring. Nurses facilitate care coordination, patient education, laboratory monitoring, transfusion administration, and timely follow-up. Dietitians provide targeted nutritional counseling to address dietary deficiencies. Laboratory professionals ensure accurate specimen handling and timely reporting of critical values. Collaborative communication among team members, with shared access to the patient’s diagnostic results and treatment plan, is essential to prevent diagnostic delay, avoid conflicting therapies, and ensure that anemia management is integrated with the treatment of comorbid conditions.
Media
(Click Image to Enlarge)
Aplastic Anemia Bone Marrow. Image demonstrating histologic findings of bone marrow biopsy in a patient with aplastic anemia.
Contributed by R Xiao, MD
(Click Image to Enlarge)
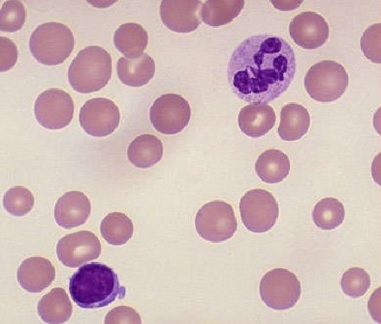
Macrocytic Anemia. Megaloblastic macrocytic anemia contains hypersegmented neutrophils and macro-ovalocytes (ie, macrocytes having an oval shape with a reduced or absent central pallor) on a peripheral blood smear.
Contributed by R Xiao, MD
(Click Image to Enlarge)
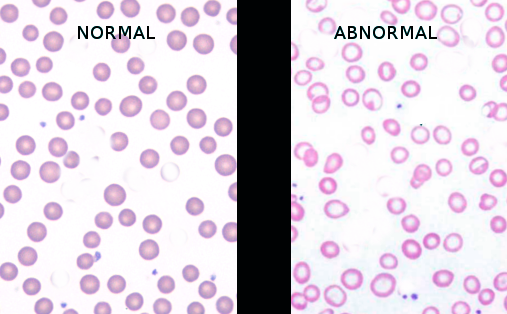
Normal Versus Iron Deficiency Anemia. The image shows a comparison of normal red blood cells and microcytic, hypochromic red blood cells in iron deficiency anemia, characterized by increased central pallor and variability in size and shape.
Contributed by S Bhimji, MD
(Click Image to Enlarge)
Sideroblastic Anemia. There are different forms of sideroblastic anemia, and all forms are defined by the presence of ring sideroblasts in the bone marrow, shown in the image.
Contributed by S Bhimji, MD
(Click Image to Enlarge)

Hypochromic Microcytic Anemia. Peripheral blood smear showing hypochromic, microcytic red blood cells with increased central pallor, consistent with a microcytic anemia pattern such as iron deficiency anemia.
Contributed by S Bhimji, MD
References
Khurana R, Kanvinde P, Mudaliar S. Revised WHO Guidelines on Hemoglobin Cutoffs to Define Anemia in Individuals and Populations. Indian pediatrics. 2024 Jul 15:61(7):671-674 [PubMed PMID: 38910369]
Jelkmann W. Regulation of erythropoietin production. The Journal of physiology. 2011 Mar 15:589(Pt 6):1251-8. doi: 10.1113/jphysiol.2010.195057. Epub 2010 Nov 15 [PubMed PMID: 21078592]
Level 3 (low-level) evidenceWeiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood. 2019 Jan 3:133(1):40-50. doi: 10.1182/blood-2018-06-856500. Epub 2018 Nov 6 [PubMed PMID: 30401705]
Hillman RS. Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. The Journal of clinical investigation. 1969 Mar:48(3):443-53 [PubMed PMID: 5773082]
Riley RS, Ben-Ezra JM, Goel R, Tidwell A. Reticulocytes and reticulocyte enumeration. Journal of clinical laboratory analysis. 2001:15(5):267-94 [PubMed PMID: 11574956]
Tefferi A. Anemia in adults: a contemporary approach to diagnosis. Mayo Clinic proceedings. 2003 Oct:78(10):1274-80 [PubMed PMID: 14531486]
GBD 2021 Anaemia Collaborators. Prevalence, years lived with disability, and trends in anaemia burden by severity and cause, 1990-2021: findings from the Global Burden of Disease Study 2021. The Lancet. Haematology. 2023 Sep:10(9):e713-e734. doi: 10.1016/S2352-3026(23)00160-6. Epub 2023 Jul 31 [PubMed PMID: 37536353]
Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet (London, England). 2018 Jan 13:391(10116):155-167. doi: 10.1016/S0140-6736(17)31822-6. Epub 2017 Jul 31 [PubMed PMID: 28774421]
Bottomley SS, Fleming MD. Sideroblastic anemia: diagnosis and management. Hematology/oncology clinics of North America. 2014 Aug:28(4):653-70, v. doi: 10.1016/j.hoc.2014.04.008. Epub 2014 Jun 2 [PubMed PMID: 25064706]
Wani AL, Ara A, Usmani JA. Lead toxicity: a review. Interdisciplinary toxicology. 2015 Jun:8(2):55-64. doi: 10.1515/intox-2015-0009. Epub [PubMed PMID: 27486361]
Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO 2026 Clinical Practice Guideline for the Management of Anemia in Chronic Kidney Disease (CKD). Kidney international. 2026 Jan:109(1S):S1-S99. doi: 10.1016/j.kint.2025.06.006. Epub [PubMed PMID: 41485812]
Level 1 (high-level) evidenceKulasekararaj A, Cavenagh J, Dokal I, Foukaneli T, Gandhi S, Garg M, Griffin M, Hillmen P, Ireland R, Killick S, Mansour S, Mufti G, Potter V, Snowden J, Stanworth S, Zuha R, Marsh J, BSH Committee. Guidelines for the diagnosis and management of adult aplastic anaemia: A British Society for Haematology Guideline. British journal of haematology. 2024 Mar:204(3):784-804. doi: 10.1111/bjh.19236. Epub 2024 Jan 21 [PubMed PMID: 38247114]
Means RT Jr. Pure red cell aplasia. Blood. 2016 Nov 24:128(21):2504-2509 [PubMed PMID: 27881371]
Silvestris F, Tucci M, Quatraro C, Dammacco F. Recent advances in understanding the pathogenesis of anemia in multiple myeloma. International journal of hematology. 2003 Aug:78(2):121-5 [PubMed PMID: 12953805]
Level 3 (low-level) evidenceGreen R, Datta Mitra A. Megaloblastic Anemias: Nutritional and Other Causes. The Medical clinics of North America. 2017 Mar:101(2):297-317. doi: 10.1016/j.mcna.2016.09.013. Epub 2016 Dec 14 [PubMed PMID: 28189172]
Aslinia F, Mazza JJ, Yale SH. Megaloblastic anemia and other causes of macrocytosis. Clinical medicine & research. 2006 Sep:4(3):236-41 [PubMed PMID: 16988104]
Stabler SP. Clinical practice. Vitamin B12 deficiency. The New England journal of medicine. 2013 Jan 10:368(2):149-60. doi: 10.1056/NEJMcp1113996. Epub [PubMed PMID: 23301732]
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, Wang SA, Bagg A, Barbui T, Branford S, Bueso-Ramos CE, Cortes JE, Dal Cin P, DiNardo CD, Dombret H, Duncavage EJ, Ebert BL, Estey EH, Facchetti F, Foucar K, Gangat N, Gianelli U, Godley LA, Gökbuget N, Gotlib J, Hellström-Lindberg E, Hobbs GS, Hoffman R, Jabbour EJ, Kiladjian JJ, Larson RA, Le Beau MM, Loh ML, Löwenberg B, Macintyre E, Malcovati L, Mullighan CG, Niemeyer C, Odenike OM, Ogawa S, Orfao A, Papaemmanuil E, Passamonti F, Porkka K, Pui CH, Radich JP, Reiter A, Rozman M, Rudelius M, Savona MR, Schiffer CA, Schmitt-Graeff A, Shimamura A, Sierra J, Stock WA, Stone RM, Tallman MS, Thiele J, Tien HF, Tzankov A, Vannucchi AM, Vyas P, Wei AH, Weinberg OK, Wierzbowska A, Cazzola M, Döhner H, Tefferi A. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022 Sep 15:140(11):1200-1228. doi: 10.1182/blood.2022015850. Epub [PubMed PMID: 35767897]
Level 3 (low-level) evidenceKhoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, Bejar R, Berti E, Busque L, Chan JKC, Chen W, Chen X, Chng WJ, Choi JK, Colmenero I, Coupland SE, Cross NCP, De Jong D, Elghetany MT, Takahashi E, Emile JF, Ferry J, Fogelstrand L, Fontenay M, Germing U, Gujral S, Haferlach T, Harrison C, Hodge JC, Hu S, Jansen JH, Kanagal-Shamanna R, Kantarjian HM, Kratz CP, Li XQ, Lim MS, Loeb K, Loghavi S, Marcogliese A, Meshinchi S, Michaels P, Naresh KN, Natkunam Y, Nejati R, Ott G, Padron E, Patel KP, Patkar N, Picarsic J, Platzbecker U, Roberts I, Schuh A, Sewell W, Siebert R, Tembhare P, Tyner J, Verstovsek S, Wang W, Wood B, Xiao W, Yeung C, Hochhaus A. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022 Jul:36(7):1703-1719. doi: 10.1038/s41375-022-01613-1. Epub 2022 Jun 22 [PubMed PMID: 35732831]
Ballard HS. The hematological complications of alcoholism. Alcohol health and research world. 1997:21(1):42-52 [PubMed PMID: 15706762]
Level 3 (low-level) evidenceSzczepanek-Parulska E, Hernik A, Ruchała M. Anemia in thyroid diseases. Polish archives of internal medicine. 2017 May 31:127(5):352-360. doi: 10.20452/pamw.3985. Epub 2017 Mar 28 [PubMed PMID: 28400547]
Scott JM, Weir DG. Drug-induced megaloblastic change. Clinics in haematology. 1980 Oct:9(3):587-606 [PubMed PMID: 6450011]
Phillips J, Henderson AC. Hemolytic Anemia: Evaluation and Differential Diagnosis. American family physician. 2018 Sep 15:98(6):354-361 [PubMed PMID: 30215915]
Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. The New England journal of medicine. 2017 Apr 20:376(16):1561-1573. doi: 10.1056/NEJMra1510865. Epub [PubMed PMID: 28423290]
Luzzatto L, Nannelli C, Notaro R. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematology/oncology clinics of North America. 2016 Apr:30(2):373-93. doi: 10.1016/j.hoc.2015.11.006. Epub [PubMed PMID: 27040960]
Jäger U, Barcellini W, Broome CM, Gertz MA, Hill A, Hill QA, Jilma B, Kuter DJ, Michel M, Montillo M, Röth A, Zeerleder SS, Berentsen S. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood reviews. 2020 May:41():100648. doi: 10.1016/j.blre.2019.100648. Epub 2019 Dec 5 [PubMed PMID: 31839434]
Level 3 (low-level) evidenceScully M, Cataland SR, Peyvandi F, Coppo P, Knöbl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F, Biswas D, De Winter H, Zeldin RK, HERCULES Investigators. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. The New England journal of medicine. 2019 Jan 24:380(4):335-346. doi: 10.1056/NEJMoa1806311. Epub 2019 Jan 9 [PubMed PMID: 30625070]
Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nature reviews. Disease primers. 2017 May 18:3():17028. doi: 10.1038/nrdp.2017.28. Epub 2017 May 18 [PubMed PMID: 28516949]
Le CH. The Prevalence of Anemia and Moderate-Severe Anemia in the US Population (NHANES 2003-2012). PloS one. 2016:11(11):e0166635. doi: 10.1371/journal.pone.0166635. Epub 2016 Nov 15 [PubMed PMID: 27846276]
Stauder R, Valent P, Theurl I. Anemia at older age: etiologies, clinical implications, and management. Blood. 2018 Feb 1:131(5):505-514. doi: 10.1182/blood-2017-07-746446. Epub 2017 Nov 15 [PubMed PMID: 29141943]
Oyedeji CI, Artz AS, Cohen HJ. How I treat anemia in older adults. Blood. 2024 Jan 18:143(3):205-213. doi: 10.1182/blood.2022017626. Epub [PubMed PMID: 36827619]
Wacka E, Nicikowski J, Jarmuzek P, Zembron-Lacny A. Anemia and Its Connections to Inflammation in Older Adults: A Review. Journal of clinical medicine. 2024 Apr 2:13(7):. doi: 10.3390/jcm13072049. Epub 2024 Apr 2 [PubMed PMID: 38610814]
O'Toole F, Sheane R, Reynaud N, McAuliffe FM, Walsh JM. Screening and treatment of iron deficiency anemia in pregnancy: A review and appraisal of current international guidelines. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics. 2024 Jul:166(1):214-227. doi: 10.1002/ijgo.15270. Epub 2023 Dec 9 [PubMed PMID: 38069617]
Pasricha SR, Tye-Din J, Muckenthaler MU, Swinkels DW. Iron deficiency. Lancet (London, England). 2021 Jan 16:397(10270):233-248. doi: 10.1016/S0140-6736(20)32594-0. Epub 2020 Dec 4 [PubMed PMID: 33285139]
Ganz T. Anemia of Inflammation. The New England journal of medicine. 2019 Sep 19:381(12):1148-1157. doi: 10.1056/NEJMra1804281. Epub [PubMed PMID: 31532961]
Rockey DC, Cello JP. Evaluation of the gastrointestinal tract in patients with iron-deficiency anemia. The New England journal of medicine. 1993 Dec 2:329(23):1691-5 [PubMed PMID: 8179652]
Ko CW, Siddique SM, Patel A, Harris A, Sultan S, Altayar O, Falck-Ytter Y. AGA Clinical Practice Guidelines on the Gastrointestinal Evaluation of Iron Deficiency Anemia. Gastroenterology. 2020 Sep:159(3):1085-1094. doi: 10.1053/j.gastro.2020.06.046. Epub 2020 Aug 15 [PubMed PMID: 32810434]
Level 1 (high-level) evidenceAnand IS, Gupta P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation. 2018 Jul 3:138(1):80-98. doi: 10.1161/CIRCULATIONAHA.118.030099. Epub [PubMed PMID: 29967232]
Allen RP, Earley CJ. The role of iron in restless legs syndrome. Movement disorders : official journal of the Movement Disorder Society. 2007:22 Suppl 18():S440-8 [PubMed PMID: 17566122]
Camaschella C. Iron-Deficiency Anemia. The New England journal of medicine. 2015 Jul 30:373(5):485-6. doi: 10.1056/NEJMc1507104. Epub [PubMed PMID: 26222573]
Sheth TN, Choudhry NK, Bowes M, Detsky AS. The relation of conjunctival pallor to the presence of anemia. Journal of general internal medicine. 1997 Feb:12(2):102-6 [PubMed PMID: 9051559]
Bouri S, Martin J. Investigation of iron deficiency anaemia . Clinical medicine (London, England). 2018 Jun:18(3):242-244. doi: 10.7861/clinmedicine.18-3-242. Epub [PubMed PMID: 29858435]
Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes's original 1934 classification to the third millennium. Current opinion in hematology. 2013 May:20(3):222-30. doi: 10.1097/MOH.0b013e32835f5933. Epub [PubMed PMID: 23449069]
Level 3 (low-level) evidenceJäger L, Rachamin Y, Senn O, Burgstaller JM, Rosemann T, Markun S. Ferritin Cutoffs and Diagnosis of Iron Deficiency in Primary Care. JAMA network open. 2024 Aug 1:7(8):e2425692. doi: 10.1001/jamanetworkopen.2024.25692. Epub 2024 Aug 1 [PubMed PMID: 39102268]
Iolascon A, Andolfo I, Russo R, Sanchez M, Busti F, Swinkels D, Aguilar Martinez P, Bou-Fakhredin R, Muckenthaler MU, Unal S, Porto G, Ganz T, Kattamis A, De Franceschi L, Cappellini MD, Munro MG, Taher A, from EHA‐SWG Red Cell and Iron. Recommendations for diagnosis, treatment, and prevention of iron deficiency and iron deficiency anemia. HemaSphere. 2024 Jul:8(7):e108. doi: 10.1002/hem3.108. Epub 2024 Jul 15 [PubMed PMID: 39011129]
DeLoughery TG, Jackson CS, Ko CW, Rockey DC. AGA Clinical Practice Update on Management of Iron Deficiency Anemia: Expert Review. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2024 Aug:22(8):1575-1583. doi: 10.1016/j.cgh.2024.03.046. Epub 2024 Jun 12 [PubMed PMID: 38864796]
Hébert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, Tweeddale M, Schweitzer I, Yetisir E. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. The New England journal of medicine. 1999 Feb 11:340(6):409-17 [PubMed PMID: 9971864]
Level 1 (high-level) evidenceHolst LB, Haase N, Wetterslev J, Wernerman J, Guttormsen AB, Karlsson S, Johansson PI, Aneman A, Vang ML, Winding R, Nebrich L, Nibro HL, Rasmussen BS, Lauridsen JR, Nielsen JS, Oldner A, Pettilä V, Cronhjort MB, Andersen LH, Pedersen UG, Reiter N, Wiis J, White JO, Russell L, Thornberg KJ, Hjortrup PB, Müller RG, Møller MH, Steensen M, Tjäder I, Kilsand K, Odeberg-Wernerman S, Sjøbø B, Bundgaard H, Thyø MA, Lodahl D, Mærkedahl R, Albeck C, Illum D, Kruse M, Winkel P, Perner A, TRISS Trial Group, Scandinavian Critical Care Trials Group. Lower versus higher hemoglobin threshold for transfusion in septic shock. The New England journal of medicine. 2014 Oct 9:371(15):1381-91. doi: 10.1056/NEJMoa1406617. Epub 2014 Oct 1 [PubMed PMID: 25270275]
Level 1 (high-level) evidenceCarson JL, Terrin ML, Noveck H, Sanders DW, Chaitman BR, Rhoads GG, Nemo G, Dragert K, Beaupre L, Hildebrand K, Macaulay W, Lewis C, Cook DR, Dobbin G, Zakriya KJ, Apple FS, Horney RA, Magaziner J, FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. The New England journal of medicine. 2011 Dec 29:365(26):2453-62. doi: 10.1056/NEJMoa1012452. Epub 2011 Dec 14 [PubMed PMID: 22168590]
Level 1 (high-level) evidenceCarson JL, Guyatt G, Heddle NM, Grossman BJ, Cohn CS, Fung MK, Gernsheimer T, Holcomb JB, Kaplan LJ, Katz LM, Peterson N, Ramsey G, Rao SV, Roback JD, Shander A, Tobian AA. Clinical Practice Guidelines From the AABB: Red Blood Cell Transfusion Thresholds and Storage. JAMA. 2016 Nov 15:316(19):2025-2035. doi: 10.1001/jama.2016.9185. Epub [PubMed PMID: 27732721]
Level 1 (high-level) evidenceMoretti D, Goede JS, Zeder C, Jiskra M, Chatzinakou V, Tjalsma H, Melse-Boonstra A, Brittenham G, Swinkels DW, Zimmermann MB. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015 Oct 22:126(17):1981-9. doi: 10.1182/blood-2015-05-642223. Epub 2015 Aug 19 [PubMed PMID: 26289639]
Stoffel NU, Cercamondi CI, Brittenham G, Zeder C, Geurts-Moespot AJ, Swinkels DW, Moretti D, Zimmermann MB. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. The Lancet. Haematology. 2017 Nov:4(11):e524-e533. doi: 10.1016/S2352-3026(17)30182-5. Epub 2017 Oct 9 [PubMed PMID: 29032957]
Level 1 (high-level) evidenceAuerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. American journal of hematology. 2016 Jan:91(1):31-8. doi: 10.1002/ajh.24201. Epub 2015 Nov 17 [PubMed PMID: 26408108]
Barcellini W, Fattizzo B. Management of autoimmune hemolytic anemia. Hematology. American Society of Hematology. Education Program. 2025 Dec 5:2025(1):305-311. doi: 10.1182/hematology.2025000719. Epub [PubMed PMID: 41347987]
Leonard A, Tisdale JF. A new frontier: FDA approvals for gene therapy in sickle cell disease. Molecular therapy : the journal of the American Society of Gene Therapy. 2024 Feb 7:32(2):264-267. doi: 10.1016/j.ymthe.2024.01.015. Epub 2024 Jan 20 [PubMed PMID: 38246166]
Kanter J, Liem RI, Bernaudin F, Bolaños-Meade J, Fitzhugh CD, Hankins JS, Murad MH, Panepinto JA, Rondelli D, Shenoy S, Wagner J, Walters MC, Woolford T, Meerpohl JJ, Tisdale J. American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood advances. 2021 Sep 28:5(18):3668-3689. doi: 10.1182/bloodadvances.2021004394C. Epub [PubMed PMID: 34581773]
Level 3 (low-level) evidenceFarmakis D, Porter J, Taher A, Domenica Cappellini M, Angastiniotis M, Eleftheriou A. 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia. HemaSphere. 2022 Aug:6(8):e732. doi: 10.1097/HS9.0000000000000732. Epub 2022 Jul 29 [PubMed PMID: 35928543]
Soma-Pillay P, Nelson-Piercy C, Tolppanen H, Mebazaa A. Physiological changes in pregnancy. Cardiovascular journal of Africa. 2016 Mar-Apr:27(2):89-94. doi: 10.5830/CVJA-2016-021. Epub [PubMed PMID: 27213856]
Hess CE, Ayers CR, Sandusky WR, Carpenter MA, Wetzel RA, Mohler DN. Mechanism of dilutional anemia in massive splenomegaly. Blood. 1976 Apr:47(4):629-44 [PubMed PMID: 1260126]
Gulati G, Uppal G, Gong J. Unreliable Automated Complete Blood Count Results: Causes, Recognition, and Resolution. Annals of laboratory medicine. 2022 Sep 1:42(5):515-530. doi: 10.3343/alm.2022.42.5.515. Epub [PubMed PMID: 35470271]
Krasowski MD. Educational Case: Hemolysis and Lipemia Interference With Laboratory Testing. Academic pathology. 2019 Jan-Dec:6():2374289519888754. doi: 10.1177/2374289519888754. Epub 2019 Nov 22 [PubMed PMID: 31803827]
Level 3 (low-level) evidenceKawai Y, Fukushima H, Asai H, Takano K, Okuda A, Tada Y, Maegawa N, Bolstad F. Significance of initial hemoglobin levels in severe trauma patients without prehospital fluid administration: a single-center study in Japan. Trauma surgery & acute care open. 2021:6(1):e000831. doi: 10.1136/tsaco-2021-000831. Epub 2021 Dec 31 [PubMed PMID: 35036573]
Henry S, Gérard D, Salignac S, Perrin J. Optimizing the management of analytical interferences affecting red blood cells on XN-10 (Sysmex®). International journal of laboratory hematology. 2022 Dec:44(6):1068-1077. doi: 10.1111/ijlh.13951. Epub 2022 Aug 22 [PubMed PMID: 36053968]
Obeid R, Andrès E, Češka R, Hooshmand B, Guéant-Rodriguez RM, Prada GI, Sławek J, Traykov L, Ta Van B, Várkonyi T, Reiners K, The Vitamin B Consensus Panelists Group. Diagnosis, Treatment and Long-Term Management of Vitamin B12 Deficiency in Adults: A Delphi Expert Consensus. Journal of clinical medicine. 2024 Apr 10:13(8):. doi: 10.3390/jcm13082176. Epub 2024 Apr 10 [PubMed PMID: 38673453]
Level 3 (low-level) evidenceSnook J, Bhala N, Beales ILP, Cannings D, Kightley C, Logan RP, Pritchard DM, Sidhu R, Surgenor S, Thomas W, Verma AM, Goddard AF. British Society of Gastroenterology guidelines for the management of iron deficiency anaemia in adults. Gut. 2021 Nov:70(11):2030-2051. doi: 10.1136/gutjnl-2021-325210. Epub 2021 Sep 8 [PubMed PMID: 34497146]
Völker LA, Brinkkoetter PT, Cataland SR, Masias C. Five years of caplacizumab - lessons learned and remaining controversies in immune-mediated thrombotic thrombocytopenic purpura. Journal of thrombosis and haemostasis : JTH. 2023 Oct:21(10):2718-2725. doi: 10.1016/j.jtha.2023.07.027. Epub 2023 Aug 9 [PubMed PMID: 37562668]
Kabrah SM. Emerging Gene Therapies in Sickle Cell Disease: A Comparative Review of Efficacy and Safety Against Standard Treatments. Journal of blood medicine. 2025:16():493-507. doi: 10.2147/JBM.S556513. Epub 2025 Nov 6 [PubMed PMID: 41220897]
Level 2 (mid-level) evidenceKremastinos DT, Farmakis D, Aessopos A, Hahalis G, Hamodraka E, Tsiapras D, Keren A. Beta-thalassemia cardiomyopathy: history, present considerations, and future perspectives. Circulation. Heart failure. 2010 May:3(3):451-8. doi: 10.1161/CIRCHEARTFAILURE.109.913863. Epub [PubMed PMID: 20484195]
Level 3 (low-level) evidenceWang R, Xu S, Hao X, Jin X, Pan D, Xia H, Liao W, Yang L, Wang S. Anemia during pregnancy and adverse pregnancy outcomes: a systematic review and meta-analysis of cohort studies. Frontiers in global women's health. 2025:6():1502585. doi: 10.3389/fgwh.2025.1502585. Epub 2025 Jan 31 [PubMed PMID: 39959784]
Level 1 (high-level) evidenceGrantham-McGregor S, Ani C. A review of studies on the effect of iron deficiency on cognitive development in children. The Journal of nutrition. 2001 Feb:131(2S-2):649S-666S; discussion 666S-668S. doi: 10.1093/jn/131.2.649S. Epub [PubMed PMID: 11160596]
Saji AM, Lui F, De Jesus O. Spinal Cord Subacute Combined Degeneration. StatPearls. 2026 Jan:(): [PubMed PMID: 32809563]
Harris AB, Badin D, Hegde V, Oni JK, Sterling RS, Khanuja HS. Preoperative Anemia is an Independent Risk Factor for Increased Complications and Mortalities After Total Knee Arthroplasty Regardless of Postoperative Transfusions. The Journal of arthroplasty. 2023 Jul:38(7 Suppl 2):S177-S181. doi: 10.1016/j.arth.2023.01.042. Epub 2023 Feb 1 [PubMed PMID: 36736931]
Ansharullah BA, Sutanto H, Romadhon PZ. Thalassemia and iron overload cardiomyopathy: Pathophysiological insights, clinical implications, and management strategies. Current problems in cardiology. 2025 Jan:50(1):102911. doi: 10.1016/j.cpcardiol.2024.102911. Epub 2024 Oct 28 [PubMed PMID: 39477176]
Connor JR, Patton SM, Oexle K, Allen RP. Iron and restless legs syndrome: treatment, genetics and pathophysiology. Sleep medicine. 2017 Mar:31():61-70. doi: 10.1016/j.sleep.2016.07.028. Epub 2016 Nov 10 [PubMed PMID: 28057495]
Camaschella C. Iron-deficiency anemia. The New England journal of medicine. 2015 May 7:372(19):1832-43. doi: 10.1056/NEJMra1401038. Epub [PubMed PMID: 25946282]