Introduction
Androgen insensitivity syndrome (AIS) is a difference (disorder) of sex development caused by impaired androgen receptor function, leading to a spectrum of phenotypic presentations. AIS represents one of the most common causes of 46,XY differences of sex development (DSD). The clinical manifestations of AIS vary with the degree of residual androgen receptor activity, ranging from a typical female phenotype in complete androgen insensitivity syndrome (CAIS) to varying degrees of undervirilization in partial androgen insensitivity syndrome (PAIS) or gynecomastia, infertility, or both in minimal androgen insensitivity syndrome (MAIS).[1]
In CAIS, complete androgen resistance results in a 46,XY individual with a typical female phenotype, whereas PAIS encompasses a broad phenotypic range depending on the extent of receptor responsiveness. The variability in presentation, particularly in PAIS, poses significant challenges in clinical treatment, including decisions regarding sex assignment at birth. A thorough understanding of androgen receptor physiology is therefore essential for accurate diagnosis, classification, and development of an appropriate treatment plan from infancy through adulthood.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Androgen insensitivity syndrome arises from loss-of-function mutations in the androgen receptor gene (AR). The androgen receptor gene is located on chromosome Xq11-12 and contains 8 exons. These mutations in the androgen receptor gene on the X chromosome result in androgen receptor dysfunction and hormone resistance. These mutations lead to a lack of virilization with or without infertility in 46,XY individuals with functional testes and adequate testosterone production. Complete and partial androgen insensitivity syndromes exhibit phenotypic variability; however, both conditions share similar genetic, endocrine, and pathophysiologic mechanisms.[2] Additionally, minimal androgen insensitivity syndrome has also been described by some authors, with no overt genital atypia and possible gynecomastia, suboptimal fertility, or both.
Epidemiology
The Office of Rare Diseases at the National Institutes of Health classifies androgen insensitivity syndrome and its subtypes as rare diseases, defined as conditions affecting fewer than 200,000 people in the US. The prevalence of individuals with a 46,XY karyotype and female phenotype is low, limiting the available data on age and clinical presentation at diagnosis in AIS. Results from a nationwide study in Denmark showed that analysis of the medical records of all known individuals with a 46, XY karyotype and female phenotype since 1960 yielded a prevalence of 6.4 per 100,000 live-born females. The prevalence of AIS was 4.1 per 100,000 live-born female individuals.[3] The prevalence of CAIS, confirmed by molecular diagnosis, is estimated to range from 1 in 20,400 to 1 in 99,100 46, XY individuals.[4]
Pathophysiology
The androgen receptor is a member of the nuclear receptor superfamily. Androgen insensitivity syndrome results from loss-of-function variants in the androgen receptor gene (AR), causing a lack of response to androgens. The AR gene has a 6-kilobase 3′ untranslated region (UTR). The gene encodes a 920-amino-acid protein that can be organized into 4 key domains: the N-terminal domain (NTD), which contains a transactivation region and ligand-independent activation function 1 (AF-1); the DNA-binding domain (DBD); a short hinge region; and a C-terminal ligand-binding domain (LBD). Another transactivation region, ligand-dependent activation function 2 (AF-2), is located in the LBD. The androgen receptor functions as a transcription factor, with AF-1 and AF-2 serving as binding sites for coactivators that modulate receptor activity (see Image. Genomic Organization and Protein Domain Architecture of the Human Androgen Receptor). More than 600 variants are known to cause the condition, distributed across all exons.[5] Relatively frequent mutations localize to the hormone-binding domain and cause androgen receptor dysfunction.[6]
The androgen receptor is kept inactive in the cytoplasm. Upon androgen binding in the LBD, the androgen receptor translocates to the nucleus and regulates transcription of its target genes. The NTD has 2 polymorphic regions, the polyglutamine (CAG) and polyglycine (GGC) repeats. Expansion of the CAG repeat beyond 38 residues leads to androgen receptor protein misfolding and spinal and bulbar muscular atrophy, also known as Kennedy disease. Spinal and bulbar muscular atrophy is a neuromuscular condition that shares some features with androgen insensitivity syndrome. CAIS represents the severe end of the phenotype, while milder phenotypes include PAIS and MAIS. The CAIS phenotype yields more variants (90% to 95%) than less severe phenotypes (approximately 30%). In addition to variants in the coding region, variants in noncoding regions, including the 5′ UTR and deep intronic regions, are now known to cause disorders within the androgen insensitivity syndrome spectrum.[7][8]
Clinical features likely depend not only on the severity of receptor dysfunction but also on other factors that contribute to varied phenotypes. In addition to noncoding region variants, non–AR gene variants affecting androgen receptor function are another possible explanation for the lower yield in the androgen insensitivity syndrome phenotype. Recently, the AR gene-variant–negative androgen insensitivity syndrome phenotype has been linked to decreased coactivator activity, leading to decreased androgen receptor function. An apolipoprotein D gene (APOD) assay has been used as a validated functional readout of androgen receptor activity in genital skin fibroblasts. Testing genital skin fibroblasts from patients with AR gene-variant–negative, clinically diagnosed androgen insensitivity syndrome showed that about one-third of the fibroblasts exhibited reduced APOD induction. Reduced APOD induction indicates diminished androgen receptor activity. This presentation has been termed androgen insensitivity syndrome type 2.[9] A formin protein and actin nucleator, disheveled-associated activator of morphogenesis 2 gene (DAAM2), and, more recently, nuclear myosin VI, have been found to play an essential role in androgen signaling and may act as coactivators affecting the androgen insensitivity syndrome phenotype.[10][11]
History and Physical
Androgen insensitivity syndrome may be subclassified into the following clinical types, primarily based on phenotype: complete androgen insensitivity syndrome (CAIS), partial androgen insensitivity syndrome (PAIS), and mild androgen insensitivity syndrome (MAIS).[12] This variability is commonly attributed to differences in residual androgen receptor activity, but the same genetic defects within a family may lead to different phenotypes. Phenotypic variability may result from differences in androgen receptor protein stability, DNA-binding capacity, and interactions with coactivators, and is more commonly observed with missense variants. More than 30 genotypes are classified under more than 1 phenotype. This variability is greater with ligand-binding domain variants and less severe phenotypes, including PAIS and MAIS, compared with CAIS. Somatic mosaicism and interindividual variability in androgen biosynthesis are other possible explanations for this variability.[13] Crosstalk between the androgen receptor gene (AR) and other genes involved in sex development, including the steroidogenic factor 1 gene (SF1) and the mitogen-activated protein kinase kinase kinase 1 gene (MAP3K1), may also affect phenotype, as described in recent reports.[14][15] In general, CAIS, PAIS, and MAIS present with primary amenorrhea, genital atypia, and infertility with or without gynecomastia, respectively. Table 1 summarizes a comparative analysis of these 3 variants.
Table 1. Comparative Analysis of Complete Androgen Insensitivity Syndrome (CAIS), Partial Androgen Insensitivity Syndrome (PAIS), and Mild Androgen Insensitivity Syndrome (MAIS)
|
Parameter |
CAIS |
PAIS |
MAIS |
|
Karyotype and sex of rearing |
46, XY; reared female |
46, XY; sex assignment depends on the degree of virilization and other factors |
46, XY; reared male |
|
Degree of androgen resistance |
Complete resistance to androgens |
Partial responsiveness, varying in degree |
Mild resistance; infrequently reported |
|
External genitalia |
Female phenotype Gonads: Abdominal or inguinal |
Undermasculinised genitalia, including micropenis, perineoscrotal (severe) hypospadias, and a bifid scrotum that may contain gonads |
Normal male; no anatomical abnormalities |
|
Internal structures |
Absent Müllerian structures (uterus, cervix, and proximal vagina), blind-ending vagina Rudimentary or absent Wolffian structures |
Absent Müllerian structures, male internal genitalia are variable, depending on the degree of androgen action. |
Normal male |
|
Typical clinical presentation |
Primary amenorrhoea in an adolescent, or inguinal swelling or hernia in infancy |
Genital ambiguity at birth warrants extensive genetic and biochemical investigation. |
Adolescent gynaecomastia or infertility presenting in later life in otherwise healthy men/boys * Bulbar and spinal muscular atrophy (Kennedy's disease) — caused by hyperexpansion of the CAG repeat (>38) in exon 1 of the AR gene |
|
Pubic and axillary hair |
Usually absent or very sparse |
Variable |
Normal |
|
Endocrine profile |
Testosterone level high-normal, LH level inappropriately elevated; FSH and inhibin levels are generally normal |
Pronounced LH and testosterone response, both basally and after hCG stimulation |
Serum LH × testosterone product proposed as a useful screening index in infertile men |
|
Fertility |
Infertile |
Generally infertile |
Possible if sperm count can be restored with high-dose androgen therapy |
|
Diagnosis |
Karyotype/phenotype mismatch (sometimes prenatal), family history of X-linked CAIS; molecular diagnosis of the androgen receptor (AR) gene. |
AR gene sequencing, ideally with an in vitro functional assay to confirm pathogenicity |
AR gene mutation analysis, including CAG repeats |
|
Key differentials |
Complete gonadal dysgenesis, Mayer-Rokitansky-Kuster-Hauser syndrome, Müllerian duct anomalies such as transverse vaginal septa, and androgen biosynthesis disorders |
Partial gonadal dysgenesis, LH receptor mutations, and androgen biosynthesis disorders |
Other causes of male infertility and gynecomastia |
Abbreviations: AR, androgen receptor gene; CAIS, complete androgen insensitivity syndrome; DSD, differences of sex development; FSH, follicle-stimulating hormone; LH, luteinizing hormone; MAIS, mild androgen insensitivity syndrome; PAIS, partial androgen insensitivity syndrome.
Evaluation
The diagnosis of complete androgen insensitivity syndrome (CAIS) and partial androgen insensitivity syndrome (PAIS) includes assessment of clinical and biochemical features, a 46, XY karyotype, and exclusion of defects in testosterone synthesis. Furthermore, genetic testing can confirm the diagnosis.
Hormonal Evaluation
Basal measurements of testosterone, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) during minipuberty in the first year of life, between 2 and 12 weeks after birth, or after puberty onset, may show high-normal testosterone levels, inappropriately increased LH levels, and normal FSH and inhibin levels. The expected postnatal surge in gonadotropins and testosterone is usually present in PAIS but may not occur in CAIS. The cause of this blunted minipuberty in these infants is not fully understood, but the lack of prenatal androgen exposure likely prevents priming of the hypothalamic-pituitary axis, resulting in a blunted LH surge.[16] However, Sertoli cell function is normal, as shown by normal inhibin B or anti-Müllerian hormone (AMH) levels. AMH levels may be elevated compared with those in unaffected infants because the androgen-insensitive state in CAIS amplifies the effect on Sertoli cells, resulting in relatively high AMH levels.[12][17] In prepubertal children, assessment of testosterone synthesis requires a human chorionic gonadotropin (hCG) stimulation test with measurement of serum androstenedione, testosterone, and dihydrotestosterone (DHT). In adults after puberty, basal hormone measurements are usually conclusive. Estradiol (E2) levels are significantly higher than the male reference range but lower than the unaffected female reference range because of aromatase-mediated conversion of testosterone to estradiol and LH-induced estradiol production by Leydig cells.
Minipuberty: At birth, all babies, regardless of 46, XY karyotype, experience a short period of gonadotropin-gonadal axis activation, known as minipuberty, around 1 to 3 months of age. During this time, the hypothalamic-pituitary axis stimulates the testes to produce testosterone by releasing LH and FSH. In CAIS, androgen receptors do not function normally; thus, a lack of prenatal hypothalamic-pituitary priming by androgens disrupts the loop, and the typical postnatal surge in gonadotropins and testosterone either fails to occur or remains blunted. In PAIS, androgen receptors function only partially; thus, testosterone feedback can still produce a postnatal rise in LH, FSH, and testosterone.
17β-Hydroxysteroid dehydrogenase type 3 deficiency: Individuals with CAIS and PAIS have normal testosterone responses after the hCG stimulation test.[18] Low serum testosterone synthesis implies impaired testosterone synthesis secondary to 17β-hydroxysteroid dehydrogenase deficiency. The testosterone-to-androstenedione ratio helps distinguish 17β-hydroxysteroid dehydrogenase deficiency from other 46,XY differences of sex development, with a ratio less than 0.8 suggestive of this condition.
Steroid 5α-reductase type 2 deficiency: Differentiation of CAIS and PAIS from this condition is made by analyzing the serum testosterone-to-DHT ratio. In patients with steroid 5α-reductase type 2 deficiency, the production of DHT decreases, and the plasma ratio of testosterone to DHT increases to greater than 20:1 to 30:1. In most children with PAIS, the production of DHT and the ratio of testosterone to DHT are normal. Individuals with CAIS may have secondary 5α-reductase type 2 deficiency due to decreased mass of urogenital tract tissues that normally produce DHT. Overlap in the testosterone to DHT ratio among some individuals with androgen insensitivity syndrome and steroid 5α-reductase type 2 deficiency makes this ratio imperfect for diagnosis, and molecular genetic analysis is often warranted. Other than baseline or stimulated testosterone with gonadotropin assays, sex hormone–binding globulin and AMH can be used as surrogate markers for adequate androgen action. AMH, a Sertoli cell marker, normally falls during puberty; AMH levels fall modestly during puberty in PAIS and do not fall in CAIS.[19]
More recently, the apolipoprotein D gene (APOD) assay has been used as a validated functional readout of androgen receptor activity in genital skin fibroblasts. Reduced APOD induction suggests diminished androgen receptor activity (see the pathophysiology section for more details).[9] These cell-based assays are particularly valuable in cases with inconclusive molecular genetic evaluation.
Importance of Genetic Testing in the Evaluation of AIS
In addition to clinical features, biochemical testing may be inadequate to establish the diagnosis of CAIS or PAIS because LH and testosterone levels overlap, especially in cases of steroid 5α-reductase type 2 deficiency. Therefore, molecular genetic studies are the preferred method for establishing the diagnosis of androgen insensitivity syndrome spectrum conditions. Results from studies showed large structural alterations along with more than 600 mutations known to cause this condition. Multiplex ligation-dependent probe amplification analysis is available to detect deletions or duplications of exons or the entire gene.[20] Sequencing of the androgen receptor gene (AR) itself may confirm the diagnosis of CAIS in 90% to 95% of cases, but only in 30% to 40% of cases with suspected PAIS. Therefore, PAIS still poses a substantial challenge for clinicians attempting to predict genotype-phenotype correlations and prognosis.
Sequencing of the entire AR gene, including the 5′ UTR and intronic regions, or whole-exome sequencing can be used to look for variants in different parts of the AR gene and non–AR genes, including mitogen-activated protein kinase kinase kinase 1 gene (MAP3K1) and nuclear receptor subfamily 5 group A member 1 gene (NR5A1), that affect phenotype. Additionally, long-read sequencing may be needed in selected cases to detect triplet-repeat disorders and assess the contributions of epigenetic changes in androgen insensitivity syndrome spectrum disorders. In addition to the AR coding region, noncoding regulatory regions, deep intronic variants, and coactivator dysfunction are implicated in androgen insensitivity syndrome spectrum disorders. They should be considered when evaluating an individual suspected of having PAIS based on clinical and hormonal evaluation with negative results on initial mutation testing.[5][21] The diagnosis of CAIS and PAIS includes assessment of clinical and biochemical features, a 46XY karyotype, and exclusion of defects in testosterone synthesis. Furthermore, genetic testing can confirm.
Treatment / Management
An interdisciplinary team comprising an endocrinologist (pediatric or adult), a urologist, a gynecologist, and a psychologist should guide treatment. Treatment of androgen insensitivity syndrome involves a holistic approach to the psychological, physiological, and social well-being of the individual with this disorder. Treatment considerations include sex of rearing in the context of genital atypia, need for hormonal replacement therapy and surgical procedures, fertility prospects, risk of tumor development if gonads are retained, and psychological assessment of gender identity, gender role, and sexual orientation. After initial assessment, individuals with androgen insensitivity syndrome require counseling and support throughout life stages. Genetic counseling is also integral to the treatment of this condition, given its genetic basis.
Complete Androgen Insensitivity Syndrome
Individuals with CAIS usually have a female sex of rearing because they have very low or no risk of gender dysphoria. CAIS may present as an incidental finding of gonads at the time of other procedures, predominantly during inguinal hernia repairs, or as primary amenorrhea in adolescents with a female phenotype. Clinicians should consider obtaining a biopsy of the gonad at the time of hernia repair and replacing the gonad subcutaneously or within the abdomen while further discussion with the parents about the diagnosis and plans for future treatment is pending.[22]
Parents can choose early gonadectomy to prevent tumorigenesis when CAIS presents in infancy, and the child is unaware of the issues surrounding the diagnosis. In this scenario, puberty induction can be performed later via estrogen replacement. Induction protocols may be similar to those used for other conditions requiring puberty induction, such as Turner syndrome, starting near 11 years of age and advancing gradually over 2.5 to 3 years. Please see StatPearls' companion reference, "Turner Syndrome," for further information. Additionally, because individuals with CAIS do not have a uterus, estrogen-induced endometrial cancer risk is absent. Therefore, unopposed estrogen therapy can be continued, unlike in other conditions.
Alternatively, gonadectomy can be delayed until early adulthood because the risk of a gonadal tumor in childhood is very low.[23][24] In this situation, puberty induction via external estrogen replacement is not needed. Puberty occurs spontaneously, manifesting as typical breast development and an appropriately timed growth spurt; however, menarche does not occur. Formal tumor surveillance is needed in individuals who choose to retain the gonads until puberty completion or later. Surgical treatment in CAIS includes vaginal dilation and, rarely, vaginoplasty for sexual function and well-being. Adoption or use of donor oocytes and a surrogate with the partner’s sperm is an option for parenthood.
Partial Androgen Insensitivity Syndrome
Unlike CAIS, infants with partial androgen insensitivity syndrome are usually born with genital atypia, which requires an accurate diagnosis and a decision on sex assignment after a holistic discussion with family and caregivers. After sex assignment, the clinician can address early treatment issues. Ongoing counseling should include discussion of expected pubertal development, fertility, gonadal tumor risk, and possible need for staged hormonal or surgical treatment.
In infants assigned male sex, medical treatment includes androgen supplementation at the time of puberty. Surgical treatment includes correction of hypospadias and undescended testes. These procedures are preferable during the second to third year of life. At the time of puberty, gynecomastia may develop, which should be corrected with reduction mammoplasty. The incidence of breast cancer in males with PAIS is low.[25]
Treatment of PAIS in infants assigned female sex includes estrogen supplementation at the time of puberty, along with genitoplasty with gonadectomy before the onset of puberty. Sometimes, puberty suppression is needed until parents and the child decide on the sex of rearing to prevent irreversible virilization. Individuals raised as male require high-dose androgen supplementation for virilization and possible fertility, though no standard regimen can be recommended for all patients. A short course of testosterone or topical dihydrotestosterone may help assess the phallus's responsiveness to androgens, and the external masculinization score at birth or presentation may be useful in predicting potential virilization during puberty and in guiding a decision regarding the sex of rearing. Surgical procedures and hormonal puberty blockade should conform to the local legal and cultural framework. Individuals with mild androgen insensitivity syndrome need treatment of gynecomastia with surgical intervention and evaluation for infertility. In addition to surgical and hormonal treatment, psychological support is needed in nearly all individuals with the androgen insensitivity syndrome spectrum.
Differential Diagnosis
The differential diagnosis of androgen insensitivity syndrome includes a range of differences in sex development that may present with undervirilization or a predominantly female phenotype in individuals with a 46, XY karyotype. These conditions can be broadly categorized into defects in androgen production and disorders of gonadal development. Complete androgen insensitivity syndrome has a more distinct phenotype and a higher yield on molecular genetic analysis, while partial androgen insensitivity syndrome has a more variable presentation and a lower yield on genetic analysis, resulting in a wider differential diagnosis.
Defects in Androgen Biosynthesis
Defects in androgen biosynthesis result from abnormalities in enzymes of the steroidogenic pathway or from dysfunction of the luteinizing hormone receptor. In these conditions, testes are present but unable to produce adequate testosterone, leading to undervirilization despite a 46, XY karyotype. Unlike the AIS spectrum, serum testosterone levels are typically low.[23] Steroid 5α-reductase type 2 deficiency is an important differential diagnosis because it presents with undervirilization and adequate testosterone levels during puberty or after human chorionic gonadotropin stimulation, unlike other androgen biosynthesis defects. 17β-Hydroxysteroid dehydrogenase type 3 deficiency is another condition that presents with female or atypical genitalia with a 46, XY karyotype, but affected individuals usually have low testosterone levels.
Disorders of Gonadal Development
Mixed gonadal dysgenesis (MGD), a chromosomal DSD, is most commonly associated with a mosaic karyotype of 45, X/46, XY. Typical findings include a dysgenetic testis on one side and a streak gonad on the other, with variable development of internal and external genitalia. Asymmetric external genitalia, with a palpable testis on one side and no palpable gonad on the other, is the typical phenotype, although presentation may vary. These individuals may share some features of Turner syndrome due to 45, X/46, XY mosaicism.
Partial gonadal dysgenesis occurs in individuals with a 46, XY karyotype and incomplete testicular development. Partial gonadal dysgenesis is characterized by reduced production of testosterone, AMH, and inhibin B, persistence of Müllerian structures, and a broad clinical spectrum ranging from ambiguous genitalia to an undervirilized male phenotype. Known genes associated with gonadal dysgenesis spectrum disorders include sex-determining region Y gene (SRY), zinc finger protein, FOG family member 2 gene (ZFPM2), nuclear receptor subfamily 5 group A member 1 gene (NR5A1), desert hedgehog signaling molecule gene (DHH), and Wnt family member 1 gene (WNT1).
Complete gonadal dysgenesis presents with absent pubertal progression and primary amenorrhea in an individual with a 46, XY karyotype. The presence of Müllerian structures in a 46, XY individual suggests impaired anti-Müllerian hormone production or action and is more consistent with gonadal dysgenesis than with AIS.
Ovotesticular DSD: In an individual with variable genital atypia with breast development, a 46, XX karyotype is more common, but a 46, XY karyotype can also occur.
Müllerian agenesis, also known as Mayer-Rokitansky-Küster-Hauser syndrome, should be considered in individuals with a female phenotype presenting with primary amenorrhea, a blind vaginal pouch, and an absent uterus.[24] The key distinction is that individuals with Müllerian agenesis have a 46, XX karyotype with normal ovarian function and normal estrogen and androgen levels. Consequently, these individuals exhibit normal pubertal development and normal axillary and pubic hair growth, unlike patients with CAIS who lack androgen action.[25]
Pertinent Studies and Ongoing Trials
In addition to genital skin fibroblast studies and androgen receptor knockout mouse studies used to elucidate androgen receptor function in affected individuals, testicular organoids originating from human induced pluripotent stem cells may be used to study the effects of androgen receptor insensitivity in a more representative model.[26]
Prognosis
Individuals with complete androgen insensitivity syndrome (CAIS) have better outcomes in terms of satisfaction with assigned sex, contentment with their body, sexual life, and overall psychological health. Individuals with suspected partial androgen insensitivity syndrome (PAIS) need more extensive hormonal, molecular, and psychological evaluation to establish a diagnosis and guide decisions regarding sex of rearing and hormonal therapy. Even after undergoing more intensive discussions and evaluation, individuals with PAIS have poorer outcomes regarding contentment with their assigned sex, irrespective of the assigned sex. Results from studies suggested an increased tumor risk greater than 30% in late adulthood if gonadectomy is not performed.[27] Boys with genetically confirmed PAIS are likely to have poorer clinical outcomes than those with 46, XY differences of sex development, normal testosterone synthesis, and no identifiable androgen receptor gene mutation.[28]
Gonadectomy is usually undertaken after puberty, except in individuals with PAIS who are raised as girls, for whom prepubertal gonadectomy is preferred to prevent irreversible virilization. However, local laws and practices should be taken into account when deciding the timing of gonadectomy. In individuals with PAIS, hormone replacement is usually required to support adequate virilization in males or feminization in females. In individuals with CAIS, estrogen replacement is also needed after gonadectomy. Urogenital surgical procedures to align external genitalia with sex of rearing may be needed in individuals with PAIS; the extent of surgical intervention varies by the degree of masculinization present.
Individuals with androgen insensitivity syndrome and cryptorchidism have an increased risk of tumorigenesis. Risk of tumors depends on 3 factors: (1) the presence of the gonadoblastoma on the Y chromosome region, candidate gene TSPY; (2) expression of the embryonic germ cell markers octamer-binding transcription factor 3/4 gene (POU5F1) or KIT ligand gene (KITLG) beyond the age of 1 year; and (3) gonad location, with higher gonad location associated with higher risk.[29] Cryptorchidism in PAIS should be corrected surgically soon after diagnosis to maintain testicular function and minimize the risk of malignant neoplasm. These tumors may be germ cell tumors or gonadoblastomas. They occur in approximately 1.5% to 2% of undescended testes and may develop into malignant neoplasms.[30]
Carcinoma in situ or intratubular germ cell neoplasia unclassified is the premalignant precursor from which tumors associated with AIS arise.[31] Carcinoma in situ arises from gonocytes or primordial germ cells and is thought to result from developmental arrest of fetal germ cells. Carcinoma in situ leads to the development of gonadoblastoma in more than 50% of cases. Individuals with CAIS are advised to have gonadectomy because of tumor risk, which may increase substantially in later adulthood.[32] Results from a meta-analysis of 15 studies and 456 patients with CAIS who underwent gonadal biopsy or surgical procedure showed that 6.14% had a premalignant lesion, with the overwhelming majority being postpubertal (82.14%). A malignant lesion was found in 1.3%, and all individuals were postpubertal.[33] The risk of germ cell tumors is higher in PAIS than in CAIS, and results from studies suggested an incidence of 15% or higher if the testes are not in a scrotal position.[34] Monitoring for carcinoma is based primarily on imaging studies. Individuals with mild androgen insensitivity syndrome need care regarding fertility and gynecomastia, and have much better outcomes compared with other variants.
Complications
Complications may present at various stages of life:
Atypical genitalia may create difficulty in deciding the sex of rearing during infancy. Later, multiple surgical procedures may be required to align the external genitalia with the sex of rearing. Complications related to these surgical procedures increase morbidity in affected individuals. Eligible individuals may also require gonadectomy. Individuals with untreated or inadequately treated androgen insensitivity syndrome and their families may face severe psychological distress during puberty and adulthood. Psychological distress is more frequent in adults with partial androgen insensitivity syndrome than in those with complete androgen insensitivity syndrome, irrespective of whether they were raised as male or female. Results from a study by Bouvattier et al showed that in men with PAIS, all aspects of sexual activity were substantially impaired.[35] Additionally, gynecomastia and infertility are common in this condition. The systemic health effects of high-dose hormonal treatment in individuals raised as male remain poorly understood. The risk of gonadal tumors varies according to the androgen insensitivity syndrome subtype and the anatomic location of the gonads.
Postoperative and Rehabilitation Care
Children diagnosed with partial androgen insensitivity syndrome and female sex of rearing need gonadectomy before puberty to avoid masculinization. In individuals with complete androgen insensitivity syndrome, gonadectomy can be performed after puberty completion because tumors in the prepubertal state are extremely uncommon, and delayed gonadectomy allows spontaneous puberty without external sex steroid supplementation. Furthermore, vaginal dilatators are an effective first-line treatment to increase the length of an existing short vagina. Vaginal surgical procedures are less commonly indicated for the creation of a functional vagina.[27] A sensitive and open approach is mandatory when discussing the postoperative care plan, taking into consideration the patient's views and the family's expectations. Psychosocial support is an integral aspect of the holistic approach to the treatment of patients with androgen insensitivity syndrome and their families.
Consultations
Pediatric and adult endocrinologists play pivotal roles in treatment, with consultations from the following departments:
- Urology
- Gynecology
- Psychology
- Neonatology
- Medical genetics
Medical social workers, pharmacists, and support groups also play an important role in improving the quality of life for affected individuals and their families.
Deterrence and Patient Education
Education of families with newborns with androgen insensitivity syndrome is vital for optimizing the care plan from the newborn period into adulthood. This goal can be achieved through an interprofessional team approach involving clinicians and support groups to address the needs of affected individuals and their families. The nurse has a central role in educating patients with AIS and their families and ensuring access to community support groups and educational materials. Medical genetics consultation and genetic counseling may help families understand the risk of recurrence in future offspring and the steps to address it. Awareness among primary care clinicians also plays an important role in timely diagnosis and optimal treatment, thereby limiting the physical and psychological morbidity associated with the condition. Clinicians must address the challenges anticipated for the family of an infant born with AIS to optimize well-being and support transition into adulthood.
Enhancing Healthcare Team Outcomes
The diagnosis of AIS carries a substantial negative impact on patients and their families. Clinicians should use an interprofessional approach when treating patients with androgen insensitivity syndrome. Treatment requires an interprofessional team of healthcare professionals, including an endocrinologist (pediatric or adult), urologist, gynecologist, primary care clinician, clinical psychologist, neonatologist, clinical geneticist, medical ethicist, physician assistant, specialty-trained nurse, and social worker. This interprofessional approach facilitates appropriate and sensitive treatment of the condition and prepares the child and family to overcome challenges, with additional support from patient advocacy groups.
Results from a study by Pasterski et al showed clinical treatment patterns for disorders of sex development, based on an online questionnaire and an audit of the DSD literature, which were sent to pediatric endocrinologists at 60 medical centers across 23 European countries. The investigators evaluated interprofessional team composition, psychological support services, and the incidence of feminizing clitoroplasty. Results from the study showed that 57% of centers regularly used an interprofessional approach, involving the services of recommended pediatric subspecialists, including a pediatric endocrinologist, pediatric surgeon or urologist, plastic surgeon, pediatric psychiatrist or psychologist, gynecologist, clinical geneticist, histopathologist, and neonatologist.[36] The interprofessional approach is also beneficial for educating healthcare professionals to interpret diagnostic tests accurately and to avoid incorrect diagnoses and inappropriate treatment across the lifespan, from infancy to adulthood.[37] Substantial opportunities remain to improve the diagnosis and treatment of all causes of 46,XY disorders of sex development, including complete and particularly partial androgen insensitivity syndrome, requiring multicenter collaboration at national and international levels.
Media
(Click Image to Enlarge)
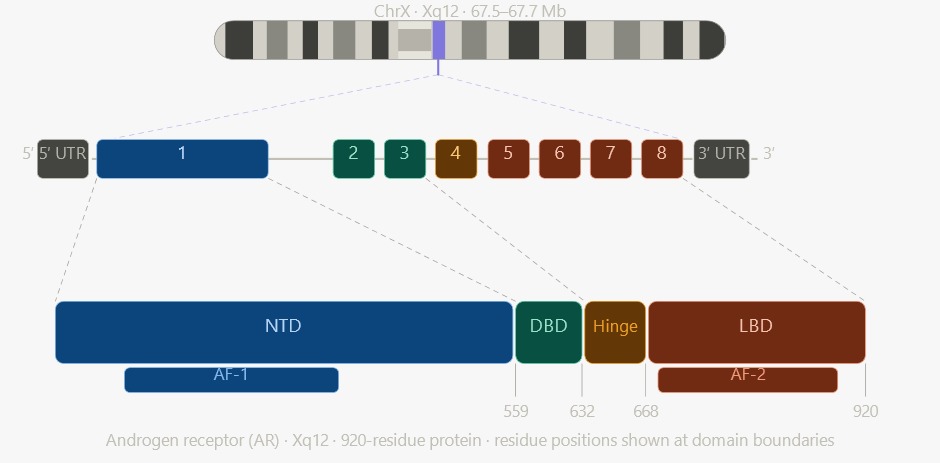
Genomic Organization and Protein Domain Architecture of the Human Androgen Receptor. The mapping of the 8-exon human androgen receptor gene at locus Xq12 to its corresponding 920-residue protein highlights the structural relationship between genomic exons and functional protein domains, including the N-terminal domain (NTD), DNA-binding domain (DBD), and ligand-binding domain (LBD). AF-1: activation function-1, and AF-2: activation function-2.
Contributed by L Sharma, MD
References
Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES Consensus Group, ESPE Consensus Group. Consensus statement on management of intersex disorders. Archives of disease in childhood. 2006 Jul:91(7):554-63 [PubMed PMID: 16624884]
Level 3 (low-level) evidenceQuigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS. Androgen receptor defects: historical, clinical, and molecular perspectives. Endocrine reviews. 1995 Jun:16(3):271-321 [PubMed PMID: 7671849]
Level 3 (low-level) evidenceBerglund A, Johannsen TH, Stochholm K, Viuff MH, Fedder J, Main KM, Gravholt CH. Incidence, Prevalence, Diagnostic Delay, and Clinical Presentation of Female 46,XY Disorders of Sex Development. The Journal of clinical endocrinology and metabolism. 2016 Dec:101(12):4532-4540 [PubMed PMID: 27603905]
Boehmer AL, Brinkmann O, Brüggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MC, Niermeijer MF, Brunner HG, Rouwé CW, Waelkens JJ, Oostdijk W, Kleijer WJ, van der Kwast TH, de Vroede MA, Drop SL. Genotype versus phenotype in families with androgen insensitivity syndrome. The Journal of clinical endocrinology and metabolism. 2001 Sep:86(9):4151-60 [PubMed PMID: 11549642]
Hornig N, Batista RL. Androgen insensitivity and the evolving genetic heterogeneity. Best practice & research. Clinical endocrinology & metabolism. 2025 Jul:39(4):102000. doi: 10.1016/j.beem.2025.102000. Epub 2025 Apr 24 [PubMed PMID: 40335402]
Werner R, Holterhus PM. Androgen action. Endocrine development. 2014:27():28-40. doi: 10.1159/000363610. Epub 2014 Sep 9 [PubMed PMID: 25247642]
Level 3 (low-level) evidenceNoveski P, Plaseski T, Dimitrovska M, Plaseska-Karanfilska D. Androgen Insensitivity Syndrome DUE to Non-Coding Variation in the Androgen Receptor Gene: Review of the Literature and Case Report of a Patient with Mosaic c.-547C}T Variant. Balkan journal of medical genetics : BJMG. 2023 Jul:26(1):51-56. doi: 10.2478/bjmg-2023-0012. Epub 2023 Jul 31 [PubMed PMID: 37576790]
Level 3 (low-level) evidenceHornig NC, de Beaufort C, Denzer F, Cools M, Wabitsch M, Ukat M, Kulle AE, Schweikert HU, Werner R, Hiort O, Audi L, Siebert R, Ammerpohl O, Holterhus PM. A Recurrent Germline Mutation in the 5'UTR of the Androgen Receptor Causes Complete Androgen Insensitivity by Activating Aberrant uORF Translation. PloS one. 2016:11(4):e0154158. doi: 10.1371/journal.pone.0154158. Epub 2016 Apr 25 [PubMed PMID: 27110943]
Hornig NC, Holterhus PM. Molecular basis of androgen insensitivity syndromes. Molecular and cellular endocrinology. 2021 Mar 1:523():111146. doi: 10.1016/j.mce.2020.111146. Epub 2020 Dec 29 [PubMed PMID: 33385475]
Knerr J, Werner R, Schwan C, Wang H, Gebhardt P, Grötsch H, Caliebe A, Spielmann M, Holterhus PM, Grosse R, Hornig NC. Formin-mediated nuclear actin at androgen receptors promotes transcription. Nature. 2023 May:617(7961):616-622. doi: 10.1038/s41586-023-05981-1. Epub 2023 Mar 27 [PubMed PMID: 36972684]
Jayawardana IM, Fleisch JM, Knerr J, Wang H, Holst M, Tholen S, Schilling O, Grosse R. Nuclear myosin VI cooperates with actin to promote transcriptional cluster formation at androgen receptors. The Journal of biological chemistry. 2026 Feb:302(2):111088. doi: 10.1016/j.jbc.2025.111088. Epub 2025 Dec 22 [PubMed PMID: 41443423]
Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. Lancet (London, England). 2012 Oct 20:380(9851):1419-28. doi: 10.1016/S0140-6736(12)60071-3. Epub 2012 Jun 13 [PubMed PMID: 22698698]
Batista RL, Rodrigues AS, Machado AZ, Nishi MY, Cunha FS, Silva RB, Costa EMF, Mendonca BB, Domenice S. Partial androgen insensitivity syndrome due to somatic mosaicism of the androgen receptor. Journal of pediatric endocrinology & metabolism : JPEM. 2018 Jan 26:31(2):223-228. doi: 10.1515/jpem-2017-0095. Epub [PubMed PMID: 29267169]
Cheng Y, Sun Y, Ji Y, Jiang D, Teng G, Zhou X, Zhou X, Li G, Xu C. Novel compound variants of the AR and MAP3K1 genes are related to the clinical heterogeneity of androgen insensitivity syndrome. Bioscience reports. 2020 May 29:40(5):. doi: 10.1042/BSR20200616. Epub [PubMed PMID: 32338288]
Naamneh Elzenaty R, Kouri C, Martinez de Lapiscina I, Sauter KS, Moreno F, Camats-Tarruella N, Flück CE. NR5A1/SF-1 Collaborates with Inhibin α and the Androgen Receptor. International journal of molecular sciences. 2024 Sep 20:25(18):. doi: 10.3390/ijms251810109. Epub 2024 Sep 20 [PubMed PMID: 39337600]
Bouvattier C, Carel JC, Lecointre C, David A, Sultan C, Bertrand AM, Morel Y, Chaussain JL. Postnatal changes of T, LH, and FSH in 46,XY infants with mutations in the AR gene. The Journal of clinical endocrinology and metabolism. 2002 Jan:87(1):29-32 [PubMed PMID: 11788616]
Hellmann P, Christiansen P, Johannsen TH, Main KM, Duno M, Juul A. Male patients with partial androgen insensitivity syndrome: a longitudinal follow-up of growth, reproductive hormones and the development of gynaecomastia. Archives of disease in childhood. 2012 May:97(5):403-9. doi: 10.1136/archdischild-2011-300584. Epub 2012 Mar 12 [PubMed PMID: 22412043]
Level 2 (mid-level) evidenceSavage MO, Chaussain JL, Evain D, Roger M, Canlorbe P, Job JC. Endocrine studies in male pseudohermaphroditism in childhood and adolescence. Clinical endocrinology. 1978 Mar:8(3):219-31 [PubMed PMID: 147759]
Josso N, Rey RA, Picard JY. Anti-müllerian hormone: a valuable addition to the toolbox of the pediatric endocrinologist. International journal of endocrinology. 2013:2013():674105. doi: 10.1155/2013/674105. Epub 2013 Dec 8 [PubMed PMID: 24382961]
Hughes IA, Werner R, Bunch T, Hiort O. Androgen insensitivity syndrome. Seminars in reproductive medicine. 2012 Oct:30(5):432-42. doi: 10.1055/s-0032-1324728. Epub 2012 Oct 8 [PubMed PMID: 23044881]
Level 3 (low-level) evidenceLiu Q, Tong Y, Wang K. Genome-wide detection of short tandem repeat expansions by long-read sequencing. BMC bioinformatics. 2020 Dec 28:21(Suppl 21):542. doi: 10.1186/s12859-020-03876-w. Epub 2020 Dec 28 [PubMed PMID: 33371889]
Deeb A, Hughes IA. Inguinal hernia in female infants: a cue to check the sex chromosomes? BJU international. 2005 Aug:96(3):401-3 [PubMed PMID: 16042738]
Almulhem B, AlJishi FM, Al-Qahtani M. Identifying 17-β-HSD3 Deficiency in Patients with Karyotype 46,XY Misdiagnosed with Androgen Insensitivity Syndrome: A Pediatric Case Report. The American journal of case reports. 2025 Oct 2:26():e948210. doi: 10.12659/AJCR.948210. Epub 2025 Oct 2 [PubMed PMID: 41035179]
Level 3 (low-level) evidenceGriffin JE, Edwards C, Madden JD, Harrod MJ, Wilson JD. Congenital absence of the vagina. The Mayer-Rokitansky-Kuster-Hauser syndrome. Annals of internal medicine. 1976 Aug:85(2):224-36 [PubMed PMID: 782313]
Level 3 (low-level) evidenceAstorino CM. Human Sexual Polymorphism and Predicted Ranges of Morphological Variation in Human Skeletal Sex Indicators. American journal of biological anthropology. 2025 Oct:188(2):e70135. doi: 10.1002/ajpa.70135. Epub [PubMed PMID: 41064945]
Nengzhuang W, Jiaming S, Minghua LIU, Long MA, Lina QIN, Xuemei GE, Hongli YAN. A brief history of testicular organoids: from theory to the wards. Journal of assisted reproduction and genetics. 2022 Jul:39(7):1423-1431. doi: 10.1007/s10815-022-02529-6. Epub 2022 Jun 2 [PubMed PMID: 35653042]
Level 2 (mid-level) evidenceAmies Oelschlager AM, Debiec K. Vaginal Dilator Therapy: A Guide for Providers for Assessing Readiness and Supporting Patients Through the Process Successfully. Journal of pediatric and adolescent gynecology. 2019 Aug:32(4):354-358. doi: 10.1016/j.jpag.2019.05.002. Epub 2019 May 12 [PubMed PMID: 31091469]
Lucas-Herald A, Bertelloni S, Juul A, Bryce J, Jiang J, Rodie M, Sinnott R, Boroujerdi M, Lindhardt Johansen M, Hiort O, Holterhus PM, Cools M, Guaragna-Filho G, Guerra-Junior G, Weintrob N, Hannema S, Drop S, Guran T, Darendeliler F, Nordenstrom A, Hughes IA, Acerini C, Tadokoro-Cuccaro R, Ahmed SF. The Long-Term Outcome of Boys With Partial Androgen Insensitivity Syndrome and a Mutation in the Androgen Receptor Gene. The Journal of clinical endocrinology and metabolism. 2016 Nov:101(11):3959-3967 [PubMed PMID: 27403927]
Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH 3rd, Mazur T, Meyer-Bahlburg HF, Mouriquand P, Quigley CA, Sandberg DE, Vilain E, Witchel S, Global DSD Update Consortium. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Hormone research in paediatrics. 2016:85(3):158-80. doi: 10.1159/000442975. Epub 2016 Jan 28 [PubMed PMID: 26820577]
Levin HS. Tumors of the testis in intersex syndromes. The Urologic clinics of North America. 2000 Aug:27(3):543-51, x [PubMed PMID: 10985153]
Rajpert-de Meyts E, Hoei-Hansen CE. From gonocytes to testicular cancer: the role of impaired gonadal development. Annals of the New York Academy of Sciences. 2007 Dec:1120():168-80. doi: 10.1196/annals.1411.013. Epub [PubMed PMID: 18184914]
Level 3 (low-level) evidenceDeans R, Creighton SM, Liao LM, Conway GS. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): patient preferences and clinical evidence. Clinical endocrinology. 2012 Jun:76(6):894-8. doi: 10.1111/j.1365-2265.2012.04330.x. Epub [PubMed PMID: 22211628]
Level 3 (low-level) evidenceBarros BA, Oliveira LR, Surur CRC, Barros-Filho AA, Maciel-Guerra AT, Guerra-Junior G. Complete androgen insensitivity syndrome and risk of gonadal malignancy: systematic review. Annals of pediatric endocrinology & metabolism. 2021 Mar:26(1):19-23. doi: 10.6065/apem.2040170.085. Epub 2021 Mar 31 [PubMed PMID: 33819955]
Level 1 (high-level) evidenceCools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocrine reviews. 2006 Aug:27(5):468-84 [PubMed PMID: 16735607]
Level 2 (mid-level) evidenceBouvattier C, Mignot B, Lefèvre H, Morel Y, Bougnères P. Impaired sexual activity in male adults with partial androgen insensitivity. The Journal of clinical endocrinology and metabolism. 2006 Sep:91(9):3310-5 [PubMed PMID: 16757528]
Pasterski V, Prentice P, Hughes IA. Consequences of the Chicago consensus on disorders of sex development (DSD): current practices in Europe. Archives of disease in childhood. 2010 Aug:95(8):618-23. doi: 10.1136/adc.2009.163840. Epub 2009 Sep 22 [PubMed PMID: 19773218]
Level 3 (low-level) evidenceBrain CE, Creighton SM, Mushtaq I, Carmichael PA, Barnicoat A, Honour JW, Larcher V, Achermann JC. Holistic management of DSD. Best practice & research. Clinical endocrinology & metabolism. 2010 Apr:24(2):335-54. doi: 10.1016/j.beem.2010.01.006. Epub [PubMed PMID: 20541156]