Introduction
Achondroplasia represents the most common genetic cause of dwarfism and the most prevalent skeletal dysplasia, characterized by severe, disproportionate short stature. This condition accounts for more than 90% of cases of disproportionate short stature (ie, dwarfism).[1] The term achondroplasia refers to impaired cartilage formation, resulting from a mutation in the transmembrane portion of fibroblast growth factor receptor-3 (FGFR3) identified between 1994 and 1995.[2] The disorder follows an autosomal dominant inheritance pattern with full penetrance, although over 80% of cases arise from spontaneous mutations. Advanced paternal age serves as a known risk factor for these de novo mutations.
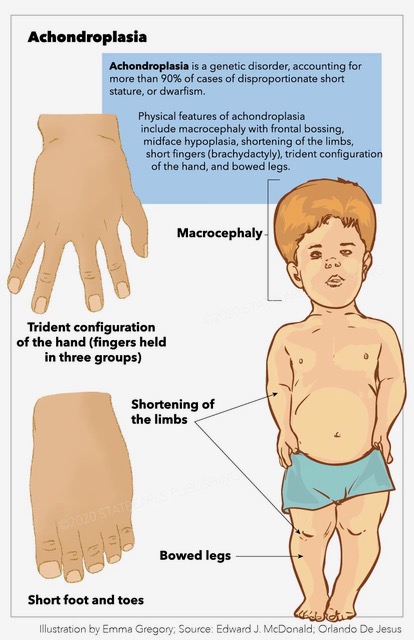
Individuals with achondroplasia generally exhibit average intelligence and have a mean lifespan approximately 10 years shorter than the general population.[3] Characteristic physical features include macrocephaly with frontal bossing, midface hypoplasia, rhizomelic shortening of the extremities, brachydactyly with a trident hand configuration, and bowed legs (genu varum).[4] The condition carries increased early childhood mortality from brainstem compression, later otolaryngologic complications, and elevated risks of obesity and cardiovascular disease in adulthood.[5][6] Affected individuals can also develop joint laxity, thoracolumbar kyphosis (TLK), and spinal stenosis that may progress and contribute to morbidity as an adult. Since 2021, vosoritide has been available as the first pharmacological precision treatment targeting the underlying pathophysiology of achondroplasia, representing a significant advance in disease-modifying therapy.[7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Achondroplasia results from a point mutation in the gene coding for the transmembrane portion of fibroblast growth factor receptor-3 (FGFR3), which resides on the short arm of chromosome 4. The resulting abnormal chondroid production affects endochondral ossification, leading to decreased linear bone growth. This pathologic process generally spares intramembranous ossification, which occurs in flat bones, eg, those in the skull (except for the skull base), face, and clavicles.
In over 80% of cases, the condition results from a sporadic (de novo) mutation. Thus, a child with achondroplasia can be born to parents with no family history of the disorder. The remaining 20% of individuals with achondroplasia have at least 1 affected parent. Achondroplasia is inherited in an autosomal dominant manner. This mutation is fully penetrant, meaning that all individuals who carry the FGFR3 heterozygous pathogenic variant exhibit the clinical manifestations of the disorder.
The remarkable homogeneity of mutations in this autosomal dominant disorder leads to a relatively low degree of heterogeneity in the achondroplasia phenotype. The risk that an offspring of an achondroplastic individual will inherit a mutated copy of the FGFR3 gene is 50%. When both parents are affected, their offspring have a 1 in 4 (25%) chance of having normal stature, a 1 in 2 (50%) chance of having achondroplasia (heterozygous), and a 1 in 4 (25%) chance of homozygous achondroplasia.[4][6] The homozygous form is usually incompatible with life, typically resulting in early neonatal death from respiratory insufficiency due to a small thoracic cage and neurologic deficits from cervicomedullary stenosis.[8]
One well-known risk factor for producing offspring with a de novo mutation is advanced paternal age, with the mutation thought to occur during spermatogenesis. Fathers aged 35 years or older have significantly increased rates of affected offspring. An increased risk of achondroplasia is not independently associated with older maternal age.[9]
Epidemiology
Achondroplasia occurs in approximately 1 in 10,000 to 1 in 30,000 live births per year. A comprehensive systematic literature review and meta-analysis published in 2020 estimated the worldwide birth prevalence of achondroplasia at 4.73 per 100,000 births (approximately 1 in 21,140 births). The global prevalence is estimated to affect approximately 1 to 9 individuals per 100,000 of the general population. The most extensive European population-based epidemiological study, published in 2019, estimated the prevalence at 3.72 per 100,000 births. This study demonstrated that prevalence remained stable over time, yet regional differences were noted. Notably, higher prevalence rates were observed in North Africa and the Middle East (34.31 per 100,000 births) and sub-Saharan Africa (12.60 per 100,000 births).[10][11] As achondroplasia follows an autosomal dominant inheritance pattern, both males and females are equally affected.
The prenatal detection rate has significantly improved in recent years. As of late 2024, noninvasive prenatal testing panels include the achondroplasia-specific FGFR3 pathogenic variants (c.1138G>A or c.1138G>C), allowing for early detection as early as 9 to 10 weeks of gestation.[12]
Pathophysiology
A point mutation in the gene coding for the transmembrane portion of FGFR3, which is located on the short arm of chromosome 4, results in the phenotypic and radiological features of achondroplasia. The point mutation arises from 2 possible base substitutions: a transition of c.1138G>A (guanine to adenine substitution identified in approximately 98% of affected individuals) and a transversion of c.1138G>C (guanine to cytosine, seen in about 1% of affected individuals). Both substitutions result in a pathogenic FGFR3 variant that leads to a gain-of-function mechanism.[13] The mutant FGFR3 protein causes constitutive activation of the receptor, resulting in excessive inhibition of chondrocyte proliferation in the growth plates of long bones.[14] This abnormal suppression of endochondral ossification leads to disproportionate shortening of the limbs and characteristic craniofacial features, including a narrow skull base. The average adult height in achondroplasia is approximately 4 feet in both sexes.[15]
History and Physical
The majority of individuals with achondroplasia receive a diagnosis in early infancy, although prenatal detection has become increasingly common. Early diagnosis proves essential, as certain complications can only be prevented through timely assessment. While formal clinical diagnostic criteria have not been published, well-established clinical and radiologic characteristics allow for accurate identification. Newborns and infants may exhibit normal overall length, yet rhizomelic shortening of the limbs serves as a hallmark feature. Craniofacial features commonly include macrocephaly with frontal and parietal bossing, a large anterior fontanelle that may persist until age 5 or 6, and midfacial retrusion resulting from underdeveloped cartilaginous facial bones. This manifests as a flattened midface, a short nasal spine, a flat nasal bridge, and an anteverted nose. Most infants display hypotonia, and combined with joint hypermobility, they often present as a “floppy baby.” Additional features include brachydactyly with a prominent gap between the ring and middle fingers (trident hand), radial head subluxation, posterior bowing of the humerus, thoracolumbar kyphosis (TLK), lumbar hyperlordosis, and genu varum, which appears in over 90% of untreated adults due to internal tibial torsion, lateral bowing, and knee laxity.[4][16][4]
Thoracolumbar kyphosis often appears in newborns and can be accentuated by a large head, trunk hypotonia, flat chest, and protuberant abdomen, particularly when infants begin to sit. The deformity may partially correct when the child lies prone. X-rays frequently show T12 and L1 apical wedging, typically involving vertebrae from T10 to L4. TLK prevalence reaches 87% in children aged 1 to 2 years and decreases to 11% by ages 5 to 10. Radiographic predictors of persistent TLK include apical vertebral translation (seen in 5% of those affected) and apical vertebral wedging for vertebral height (seen in 6% of those affected).[17] Developmental delays in gross motor skill acquisition (compared with other children with achondroplasia) are highly associated with the progression of thoracolumbar kyphosis.[3]
Foramen magnum stenosis is usually the first manifestation observed in infants with achondroplasia. Increased vigilance is required in both the evaluation and surveillance of this condition, as it is associated with a high mortality rate, ranging from 2% to 5%. Common signs and symptoms include periods of sleep apnea and excessive snoring. Other subjective or objective findings include difficulty swallowing, lower cranial nerve palsies, hyperreflexia, generalized hypotonia, weakness, and clonus. Intelligence is usually average unless the child develops hydrocephalus or has other central nervous system complications. Some infants will also develop hydrocephalus, thought to be due to increased intracranial venous pressure secondary to stenosis of the jugular foramina or to fourth ventricular outlet obstruction.[18]
Hearing impairment arises from middle ear dysfunction, and inadequate screening or treatment may lead to conductive hearing loss severe enough to affect language development. Approximately 50% of children require pressure-equalizing tympanostomy and ear tube placement, with around 40% experiencing functionally significant hearing loss.[19][20][21] Dental and orthodontic issues, including anterior crossbite, open bite, and crowding or misalignment, frequently occur in children, adolescents, and adults.[22][23]
Most individuals maintain normal intelligence, but short stature may contribute to psychosocial challenges, often beginning in childhood and worsening with age.[24] Later development usually involves pseudoclaudication, leg paresthesias, discomfort while standing, difficulty walking for prolonged periods, and subjective weakness, largely resulting from spinal stenosis (see Image. Achondroplasia). Achondroplastic patients commonly develop cervical and lumbar stenosis due to short pedicles, thickened facets, and hypertrophied ligamentum flavum. A Norwegian study reported symptomatic spinal stenosis in 68% of adults, with a median age of onset at 33 years. Lumbar spinal stenosis prevalence ranges from 25% to 68%, with roughly one-third requiring surgical intervention during their lifetime.[25][26][27][28]
Metabolic disorders—including diabetes mellitus, hypercholesterolemia, hypertension, obesity, and low bone mineral density—occur with increased frequency in individuals with achondroplasia, necessitating comprehensive monitoring and preventive care.[26]
Evaluation
Diagnostic algorithms for achondroplasia have not been established; however, diagnosis can generally be made based on characteristic clinical features and specific radiologic findings. Confirmation can be achieved through molecular genetic testing for the FGFR3 mutation. FGFR3 testing is recommended for children presenting atypically or to differentiate achondroplasia from similar skeletal dysplasias. Multigene panels, including FGFR3 and other relevant genes, may be utilized to determine the specific form of skeletal dysplasia.
Prenatal testing is indicated when there is a positive family history, given the autosomal dominant inheritance pattern. Noninvasive prenatal testing using cell-free fetal DNA from maternal serum can detect FGFR3 mutations as early as 9 to 10 weeks of gestation. A second-trimester ultrasound around 22 weeks can identify hallmark sonographic features, eg, femoral bowing and rhizomelia.[29] When achondroplasia is suspected prenatally, a consultation before delivery with a skeletal dysplasia expert, pediatrician, fetal center, geneticist, or pediatric endocrinologist is recommended to provide anticipatory guidance, outline routine surveillance, discuss specialists involved in care, review available treatments starting at birth, share outcome data, and present ongoing clinical trials.[12] Preimplantation genetic diagnosis now offers options for parents pursuing in vitro fertilization and embryo implantation.
Skeletal surveys in children with achondroplasia often reveal a contracted skull base, rhizomelic shortening of long bones, proximal femoral radiolucency, generalized metaphyseal flaring, inverted V-shaped distal femoral epiphyses, and a “champagne-glass” shaped pelvis with a wide outlet and small sacrosciatic notch. Spinal imaging typically shows narrowed interpedicular distances, short pedicles from L1 to S1, vertebral body wedging (commonly at T12 or L1), and generalized posterior vertebral scalloping.[30]
If signs of sleep apnea or myelopathy appear, sleep studies and advanced imaging of the cervicomedullary junction—eg, computed tomography (CT) or magnetic resonance imaging (MRI)—may be indicated. The Achondroplasia Foramen Magnum Score (AFMS) provides standardized MRI interpretation for screening foramen magnum stenosis, demonstrating good interobserver reliability (ICC 0.72) and guiding surgical decision-making.[31][32] The European Achondroplasia Forum recommends clinical monitoring every 3 to 4 months from birth to 1 year, then every 3 to 6 months until age 3, alongside routine MRI at 3 to 6 months and repeated as clinically indicated.[33]Cine MRI studies can further guide management, especially in severe stenosis, where craniospinal fluid (CSF) flow ceases at the cervicomedullary junction. Some authors recommend dynamic cervical MRIs in cases of symptomatic patients unless there is already complete obliteration of CSF flow in the neutral position.[27] When obtaining MRIs to evaluate for foramen magnum stenosis, studies have shown that not only is the width of the foramen magnum important, but also the relationship between the dens and opisthion, as well as the posterior tilt of C1 to C2.[34]
During each health supervision visit, including at birth, total body length, weight, and occipitofrontal circumference should be measured and plotted on achondroplasia-specific growth charts. Ongoing evaluation and care should follow established guidelines, including the American Academy of Pediatrics health supervision recommendations and the International Consensus Statement on diagnosis, interprofessional management, and lifelong care of achondroplasia.[6][35]
|
Pause and Reflect |
A 6-month-old infant with genetically confirmed achondroplasia presents for routine follow-up. The parents report that the baby has been snoring loudly at night and occasionally seems to stop breathing for brief periods during sleep. On examination, the infant has normal tone and reflexes, is meeting developmental milestones appropriately, and has a head circumference at the 97th percentile for achondroplasia-specific growth curves. The pediatrician is considering the next steps for evaluation.
|
Treatment / Management
Management of achondroplasia involves an interprofessional team approach, and anticipatory care is essential. Multiple versions of health supervision and treatment guidelines exist for those with achondroplasia, with the most comprehensive being the International Consensus Statement published in 2022, which provides detailed examinations and specific anticipatory guidance for each age group across the lifespan. These guidelines should be followed closely by the pediatrician and other health practitioners. Close attention should be paid to the development of obesity in these children and monitored to avoid complications later in life. Treatment is directed against the specific issues encountered in achondroplasia.[6][35](B3)
Macrocephaly
Distinguishing between a normal increase in occipitofrontal circumference (OFC) and an abnormal rise that may indicate clinically significant hydrocephalus proves essential in patients with achondroplasia. Head circumference should be measured at every healthcare visit and plotted on achondroplasia-specific growth charts to allow early detection of potential concerns.
Delayed skull sutural maturation in achondroplasia necessitates continuing cranial circumference measurements until age 6. Hydrocephalus requiring intervention occurs in approximately 5% of cases, with foramen magnum stenosis often contributing to its development. Surgical management options include endoscopic third ventriculostomy,[36][37] ventricular shunting, or posterior fossa decompression to relieve obstruction and prevent neurological complications.[38][36][39](A1)
Foramen Magnum Stenosis
For patients with foramen magnum stenosis, current international guidelines recommend a structured approach to screening and management. The American Academy of Pediatrics and European Achondroplasia Forum recommend obtaining polysomnography and MRI in infants with achondroplasia to evaluate for foramen magnum stenosis, rather than relying on clinical examination alone, given its low sensitivity (28%) for severe stenosis.[33][40]
The Achondroplasia Foramen Magnum Score (AFMS) provides a standardized MRI assessment, grading stenosis from 0 (no stenosis with preservation of CSF spaces) to 4 (severe stenosis with cord signal change). Studies demonstrate good interobserver reliability (ICC 0.72) for the AFMS.[31][32]
Management of foramen magnum stenosis involves routine clinical surveillance of all infants and young children, MRI screening with AFMS scoring during infancy, and referral to neurosurgery for cases with AFMS grades 3 to 4 or concerning clinical findings. Clinical examination, polysomnography, and MRI results should be considered to guide surgical decision-making, with a joint approach involving families when discussing potential decompression surgery.[33]
Treatment is surgical with foramen magnum decompression and upper cervical laminectomy with or without duraplasty. A recent 15-year single-center study found that 65% of children with achondroplasia in the cohort underwent foramen magnum decompression, with surgery improving the severity of foramen magnum stenosis and showing a nonsignificant trend toward decreased central apnea.[40]
Complete resolution or partial improvement in preoperative symptoms is usually obtained. Children with foramen magnum stenosis identified on imaging studies should avoid activities that place them at risk of head or neck injury.
Hearing Impairment
Beyond newborn screening, audiologic assessments should be completed by age 1 and repeated annually until school age, continuing annually if tympanostomy tubes are in place. Children with achondroplasia who experience recurrent otitis media or hearing difficulties should be referred to a pediatric otolaryngologist. Long-term tympanostomy tubes are often needed and may be required until around 8 years of age.[21] Additonally, speech therapy should be provided when hearing deficits are detected during routine audiological screening.(B2)
Sleep Apnea
Obstructive sleep apnea (OSA) of noncentral etiology needs to be ruled out and treated by a pediatric otolaryngologist. Children with severe OSA may require weight reduction, adenotonsillectomy, positive airway pressure at night, and tracheostomy for very severe cases.[41]
Thoracolumbar Kyphosis
TLK often presents in the newborn and usually resolves spontaneously by 18 months of age in approximately 90% of affected children, paralleling the child's ability to ambulate as the trunk muscles strengthen. In achondroplastic children younger than 1 year, unsupported sitting should be limited to help prevent the progression of kyphosis.[6][42](A1)
Bracing may be indicated if TLK progresses to a fixed deformity greater than 30 degrees (measured when prone) and when an anterior vertebral wedging or posterior displacement of vertebrae at the apex of deformity is noted. Continuation of bracing is recommended until the child is walking independently, the anterior corners of the vertebrae reconstitute, and the fixed component of the curve shows no further improvement. As bracing in these children may be poorly tolerated, an anterior and posterior fusion can be performed for a residual sagittal deformity greater than 60 degrees by age 5.[43]
Surgery should be reserved for those with neurologic deficits or fixed kyphosis measuring greater than 50 degrees. A compensatory mechanism of hyperlordosis at the lumbosacral level after age 3 plays a role, as TLK is shown to decrease gradually until age 10.[44]
Lumbar Spinal Stenosis
Lumbar spinal stenosis is the most likely cause of disability later in life and represents a significant burden in the adult achondroplasia population. A Norwegian population-based study published in 2020 found that 68% of adults with achondroplasia developed symptomatic spinal stenosis, with a median age at onset of 33 years (range 10-67 years).[25]
Evaluation for stenosis with x-rays should be considered in all patients wishing to play contact sports or participate in high-risk activities. Lumbar stenosis may first be treated nonoperatively with modalities, eg, weight loss, physical therapy, and corticosteroid injections.
Operative intervention is indicated when progressive symptoms, eg, severe claudication, urinary retention, and neurologic symptoms, are present. Early surgical intervention is important, as time from symptom onset to surgery is a significant predictor of long-term functional outcome. Patients who underwent surgery within 6 months of symptom onset were 7.13 times more likely to improve walking distance and 4 times more likely to improve functional status compared to those with delays greater than 6 months.[45][46]
Surgical treatment includes a multilevel laminectomy, including lateral recess decompression. These data suggest that wide decompression with long fusion and the use of interbody cages at the caudal level should be considered to reduce the risk of revision.[47] Complications of spinal surgery in achondroplasia include higher rates of dural tears (ranging from 5% to 55% in adults) compared to the general population, likely due to anatomic sequelae of the underlying dysplasia.[48]
Bowed Legs
Assessment for leg bowing and internal tibial torsion should be included in physical examination, and any abnormalities should prompt referral to an orthopedic specialist. Surgical indications can be challenging to determine, but may include leg or knee pain, gait abnormalities (eg, lateral thrust), or significant deformity. Patients with progressive or symptomatic bowing may benefit from corrective procedures, eg, hemiepiphysiodesis or tibial and fibular osteotomies, with or without accompanying femoral osteotomies.[12]
Obesity Management
Obesity represents a major health concern in achondroplasia, with studies showing that 67% of adults have a body mass index (BMI) in the obesity range (greater than or equal to 30 kg/m²). Individuals with achondroplasia are predisposed to abdominal obesity, particularly visceral adiposity. A 2021 study examining cardiovascular risk factors and body composition found that, despite a high BMI, adults with achondroplasia had lower blood pressure, lower total and LDL cholesterol, lower triglycerides, and significantly lower visceral fat stores (1.9 L versus 5.3 L in controls) compared with BMI-matched average-stature controls. Only individuals with very high BMI (greater than or equal to 43 kg/m²) and waist circumferences greater than or equal to 107 cm developed type 2 diabetes in this cohort.[49][50]
These findings suggest that standard BMI cutoffs may not appropriately reflect cardiovascular risk in achondroplasia. Nevertheless, obesity management remains important as excessive weight exacerbates skeletal symptoms, including lumbar spine issues, joint pain, lower limb deformity, sleep apnea, and functional limitations.
Management strategies focus on the early identification of children at high risk for obesity, along with promoting a healthy diet and regular physical activity tailored to each child’s physical abilities. Notably, the mean energy intake in adults with achondroplasia is only about 10% higher than their resting energy expenditure, indicating that obesity in this population is not solely related to excessive caloric intake. An interprofessional approach incorporating nutritional guidance, physical therapy, and ongoing medical monitoring is recommended to support healthy growth and weight management.
Pharmacological Therapy
Vosoritide represents the first precision pharmacological treatment for achondroplasia, targeting the underlying pathophysiology. Vosoritide is a C-type natriuretic peptide analogue that inhibits the downstream signaling pathway of FGFR3, which is constitutively activated in achondroplasia. The FDA initially approved vosoritide on November 19, 2021, for children older than 5 years with open epiphyses. The indication was subsequently expanded on October 20, 2023, to include treatment starting at birth.[12][51][52](A1)
|
Pause and Reflect |
A 32-year-old woman with achondroplasia presents with a 6-month history of worsening lower back pain and leg numbness that limits her ability to walk more than 1 block. Her BMI is 35 kg/m². MRI reveals multilevel lumbar spinal stenosis. She asks about her cardiovascular risk given her obesity and whether surgery would help her symptoms.
|
Differential Diagnosis
Over 350 skeletal dysplasias are known to cause short stature, most of which are very rare. However, only a few conditions exist that may be confused with achondroplasia. Hypochondroplasia and thanatophoric dysplasia are 2 other disorders with features of rhizomelic dwarfism. Both arise from a similar genetic defect as achondroplasia, with different pathogenic variants in FGFR3 that result in differing levels of FGFR3 activation, and both should be included in the differential diagnosis. FGFR3 molecular genetic testing should always be performed in children with an atypical presentation or when differentiation from similar disorders is required.[53][54]
Hypochondroplasia
The distinction between achondroplasia and hypochondroplasia is usually very difficult to make, as most of the radiologic and clinical features overlap. Patients affected by this disorder appear normal at birth. However, their arms and legs do not develop properly, and their bodies become thicker and shorter than normal. Hypochondroplasia is caused by a defect in the FGFR3 at chromosome 4 at the 4p16.3 area. A smaller height difference is noted than in achondroplasia. Approximately 10% of cases have mild intellectual disability.[55]
Thanatophoric Dysplasia
Thanatophoric dysplasia causes a form of short-limb dwarfism and is typically lethal in the perinatal period. Thanatophoric dysplasia has 2 clinically distinct forms, type I and type II. Type I is characterized by micromelia with bowed femurs. Type II is characterized by micromelia with straight femurs and uniform presence of moderate-to-severe cloverleaf skull deformity. Varying severities of the cloverleaf skull can also be present in type I. Both types share features, including infantile hypotonia, macrocephaly, frontal bossing, flat facies with ocular proptosis, brachydactyly, and micromelia. Most infants with this disorder die due to respiratory insufficiency shortly following birth.
The condition is usually incidentally discovered and suspected during a routine prenatal ultrasound, with characteristic findings of shortened long bones visible as early as 14 weeks of gestation. Specific findings during second and third-trimester ultrasounds include a cloverleaf skull, macrocephaly, ventriculomegaly, increased nuchal translucency, a narrow chest cavity with short ribs, and bowed femurs (hanatophoric dysplasia type I).[56] Thanatophoric dysplasia is also caused solely by mutations in the FGFR3 gene, and the pathogenic variant p.Lys650Glu has been identified in all individuals diagnosed with thanatophoric dysplasia type II.[57]
Severe Achondroplasia With Developmental Delay and Acanthosis Nigricans
Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) is another dysplastic condition caused by a different heterozygous mutation in the gene encoding FGFR3 on chromosome 4p16. These individuals can develop extensive areas of acanthosis nigricans starting in early childhood and suffer from severe neurological impairments. Investigations of SADDAN revealed that the mutation resulted in severe disturbances in endochondral bone growth, which approached the severity seen in thanatophoric dysplasia type I. Osseous deformities seen included femoral bowing, apex posterior tibial and fibular bowing, and curved "ram's horn" deformities of the clavicles.[58]
Acanthosis nigricans can also be seen in those with other FGFR3 genetic disorders. Therefore, the progressive skin changes should be considered a long-term complication rather than a specific feature of SADDAN.[59]
Pseudoachondroplasia
Pseudoachondroplasia, a clinically and genetically distinct skeletal dysplasia, may also appear similar. This condition also follows an autosomal dominant pattern of inheritance, yet is caused by a defect in cartilage oligomeric matrix protein, located on chromosome 19.
Signs and symptoms of this disorder include cervical instability and scoliosis with increased lumbar lordosis and significant lower extremity bowing. Individuals with this condition can develop hip, knee, and elbow flexion contractures, as well as precocious osteoarthritis. Radiographic findings include metaphyseal flaring and delayed epiphyseal ossification.[60]
Prognosis
Those diagnosed with this disorder have increased mortality rates in childhood. Multiple studies have demonstrated that the osseous abnormalities seen in achondroplastic patients, eg, foramen magnum stenosis and spinal stenosis, contribute to the significant effect on morbidity and mortality seen in all age groups, but mainly affect the pediatric group who present with foramen magnum stenosis. The mean lifespan of patients with achondroplasia has been estimated to be 61 years, approximately 10 years shorter than that of the general population.[26][61]
Complications
A diagnosis of achondroplasia involves numerous medical complications that require careful monitoring throughout life. Common complications include hydrocephalus and a range of otolaryngologic issues, eg, recurrent otitis media, hearing loss, and obstructive sleep apnea. Rapid weight gain during infancy often contributes to childhood obesity, which can exacerbate other health concerns. Surgical interventions, while sometimes necessary, carry the potential for additional morbidity and demand careful planning and follow-up. Comprehensive management of achondroplasia requires ongoing surveillance, early recognition of complications, and coordination across multiple specialties to optimize patient outcomes and minimize long-term risks.
Consultations
Given the wide range of manifestations, management of the disease requires an interprofessional team that includes consultation with a pediatrician, medical geneticist, pediatric endocrinologist, orthopedic surgeon, audiologist, otolaryngologist, pulmonologist or sleep specialist, neurologist, neurosurgeon, physical therapist, nutritionist or dietitian, and psychology and social work support.
Deterrence and Patient Education
Achondroplasia accounts for more than 90% of cases of dwarfism. Although the condition follows an autosomal dominant inheritance pattern with full penetrance, over 80% of cases result from spontaneous mutations. Genetic counseling is recommended for patients and families to discuss inheritance patterns, recurrence risks, and family planning options. Achondroplasia carries an increased risk of early childhood mortality, later otolaryngologic complications, and a higher likelihood of obesity and related comorbidities in adulthood.
Affected individuals experience a wide range of medical complications that require careful evaluation and ongoing surveillance. Joint laxity, thoracolumbar kyphosis, and spinal stenosis can progress over time and contribute to adult morbidity. Foramen magnum stenosis often presents first in infancy and demands close monitoring. Lumbar spinal stenosis develops in approximately 25% of individuals with achondroplasia, frequently affecting mobility and quality of life. Effective management relies on an interprofessional team and anticipatory care, ensuring timely interventions, surveillance, and coordination across specialties. Life expectancy for patients with achondroplasia is slightly shorter than that of the general population, underscoring the importance of proactive medical management.
Enhancing Healthcare Team Outcomes
Achondroplasia is the most common genetic cause of disproportionate short stature, resulting from FGFR3 mutations that impair endochondral ossification. Affected individuals present with characteristic features including macrocephaly, midface hypoplasia, rhizomelic limb shortening, thoracolumbar kyphosis, spinal stenosis, and increased risk of obesity and cardiometabolic complications. Early identification, accurate diagnosis through clinical, radiographic, and molecular assessment, and anticipatory care are critical to prevent severe complications such as foramen magnum stenosis, obstructive sleep apnea, hearing loss, and progressive orthopedic deformities. The availability of disease-modifying therapy, such as vosoritide, emphasizes the importance of early and ongoing surveillance, multidisciplinary management, and family education to optimize outcomes across the lifespan.
Management requires an interprofessional approach integrating pediatricians, geneticists, otolaryngologists, endocrinologists, neurologists, neurosurgeons, orthopedic surgeons, dentists, audiologists, psychologists, and rehabilitation specialists. Clinicians coordinate care by implementing standardized surveillance protocols, monitoring growth and neurologic status, guiding safe physical activity, and addressing psychosocial needs. Effective communication, anticipatory guidance, and early intervention strategies—including surgery, pharmacologic therapy, and physical therapy—enhance patient safety, functional independence, and quality of life. Engagement with support networks, adaptive resources, and education on mobility and social participation further supports holistic, patient-centered care.
Media
(Click Image to Enlarge)
Achondroplasia. Clinical features of achondroplasia include trident configuration, macrocephaly, bowed legs, dwarfism, and frontal bossing.
StatPearls Publishing Illustration
References
Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994 Jul 29:78(2):335-42 [PubMed PMID: 7913883]
Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano CA. Achondroplasia is defined by recurrent G380R mutations of FGFR3. American journal of human genetics. 1995 Feb:56(2):368-73 [PubMed PMID: 7847369]
Wynn J, King TM, Gambello MJ, Waller DK, Hecht JT. Mortality in achondroplasia study: a 42-year follow-up. American journal of medical genetics. Part A. 2007 Nov 1:143A(21):2502-11 [PubMed PMID: 17879967]
Pauli RM. Achondroplasia: a comprehensive clinical review. Orphanet journal of rare diseases. 2019 Jan 3:14(1):1. doi: 10.1186/s13023-018-0972-6. Epub 2019 Jan 3 [PubMed PMID: 30606190]
Hashmi SS, Gamble C, Hoover-Fong J, Alade AY, Pauli RM, Modaff P, Carney M, Brown C, Bober MB, Hecht JT. Multicenter study of mortality in achondroplasia. American journal of medical genetics. Part A. 2018 Nov:176(11):2359-2364. doi: 10.1002/ajmg.a.40528. Epub 2018 Oct 1 [PubMed PMID: 30276962]
Level 2 (mid-level) evidenceHoover-Fong J, Scott CI, Jones MC, COMMITTEE ON GENETICS. Health Supervision for People With Achondroplasia. Pediatrics. 2020 Jun:145(6):. pii: e20201010. doi: 10.1542/peds.2020-1010. Epub [PubMed PMID: 32457214]
Savarirayan R, Hoover-Fong J, Ozono K, Backeljauw P, Cormier-Daire V, DeAndrade K, Ireland P, Irving M, Llerena Junior J, Maghnie M, Menzel M, Merchant N, Mohnike K, Iruretagoyena SN, Okada K, Fredwall SO. International consensus guidelines on the implementation and monitoring of vosoritide therapy in individuals with achondroplasia. Nature reviews. Endocrinology. 2025 May:21(5):314-324. doi: 10.1038/s41574-024-01074-9. Epub 2025 Jan 6 [PubMed PMID: 39757323]
Level 3 (low-level) evidenceHall JG. The natural history of achondroplasia. Basic life sciences. 1988:48():3-9 [PubMed PMID: 3071358]
Waller DK, Correa A, Vo TM, Wang Y, Hobbs C, Langlois PH, Pearson K, Romitti PA, Shaw GM, Hecht JT. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. American journal of medical genetics. Part A. 2008 Sep 15:146A(18):2385-9. doi: 10.1002/ajmg.a.32485. Epub [PubMed PMID: 18698630]
Foreman PK, van Kessel F, van Hoorn R, van den Bosch J, Shediac R, Landis S. Birth prevalence of achondroplasia: A systematic literature review and meta-analysis. American journal of medical genetics. Part A. 2020 Oct:182(10):2297-2316. doi: 10.1002/ajmg.a.61787. Epub 2020 Aug 17 [PubMed PMID: 32803853]
Level 1 (high-level) evidenceCoi A, Santoro M, Garne E, Pierini A, Addor MC, Alessandri JL, Bergman JEH, Bianchi F, Boban L, Braz P, Cavero-Carbonell C, Gatt M, Haeusler M, Klungsøyr K, Kurinczuk JJ, Lanzoni M, Lelong N, Luyt K, Mokoroa O, Mullaney C, Nelen V, Neville AJ, O'Mahony MT, Perthus I, Rankin J, Rissmann A, Rouget F, Schaub B, Tucker D, Wellesley D, Wisniewska K, Zymak-Zakutnia N, Barišić I. Epidemiology of achondroplasia: A population-based study in Europe. American journal of medical genetics. Part A. 2019 Sep:179(9):1791-1798. doi: 10.1002/ajmg.a.61289. Epub 2019 Jul 11 [PubMed PMID: 31294928]
Merchant N, Hoover-Fong J, Carroll RS. Approach to the Patient with Achondroplasia-New Considerations for Diagnosis, Management, and Treatment. The Journal of clinical endocrinology and metabolism. 2025 Jun 17:110(7):e2309-e2316. doi: 10.1210/clinem/dgaf017. Epub [PubMed PMID: 39813116]
Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. The EMBO journal. 1996 Feb 1:15(3):520-7 [PubMed PMID: 8599935]
Bouali H, Latrech H. Achondroplasia: Current Options and Future Perspective. Pediatric endocrinology reviews : PER. 2015 Jun:12(4):388-95 [PubMed PMID: 26182483]
Level 3 (low-level) evidenceHorton WA, Rotter JI, Rimoin DL, Scott CI, Hall JG. Standard growth curves for achondroplasia. The Journal of pediatrics. 1978 Sep:93(3):435-8 [PubMed PMID: 690757]
Inan M, Thacker M, Church C, Miller F, Mackenzie WG, Conklin D. Dynamic lower extremity alignment in children with achondroplasia. Journal of pediatric orthopedics. 2006 Jul-Aug:26(4):526-9 [PubMed PMID: 16791073]
Level 2 (mid-level) evidenceBorkhuu B, Nagaraju DK, Chan G, Holmes L Jr, Mackenzie WG. Factors related to progression of thoracolumbar kyphosis in children with achondroplasia: a retrospective cohort study of forty-eight children treated in a comprehensive orthopaedic center. Spine. 2009 Jul 15:34(16):1699-705. doi: 10.1097/BRS.0b013e3181ac8f9d. Epub [PubMed PMID: 19770611]
Level 2 (mid-level) evidenceReid CS, Pyeritz RE, Kopits SE, Maria BL, Wang H, McPherson RW, Hurko O, Phillips JA 3rd, Rosenbaum AE. Cervicomedullary compression in young patients with achondroplasia: value of comprehensive neurologic and respiratory evaluation. The Journal of pediatrics. 1987 Apr:110(4):522-30 [PubMed PMID: 3559799]
Ireland PJ, Donaghey S, McGill J, Zankl A, Ware RS, Pacey V, Ault J, Savarirayan R, Sillence D, Thompson E, Townshend S, Johnston LM. Development in children with achondroplasia: a prospective clinical cohort study. Developmental medicine and child neurology. 2012 Jun:54(6):532-7. doi: 10.1111/j.1469-8749.2012.04234.x. Epub 2012 Mar 12 [PubMed PMID: 22409389]
Level 2 (mid-level) evidenceTunkel DE, Kerbavaz R, Smith B, Rose-Hardison D, Alade Y, Hoover-Fong J. Hearing screening in children with skeletal dysplasia. Archives of otolaryngology--head & neck surgery. 2011 Dec:137(12):1236-9. doi: 10.1001/archoto.2011.206. Epub [PubMed PMID: 22183904]
Berkowitz RG, Grundfast KM, Scott C, Saal H, Stern H, Rosenbaum K. Middle ear disease in childhood achondroplasia. Ear, nose, & throat journal. 1991 May:70(5):305-8 [PubMed PMID: 1914954]
Level 2 (mid-level) evidencePfeiffer KM, Brod M, Smith A, Viuff D, Ota S, Charlton RW. Functioning and well-being in older children and adolescents with achondroplasia: A qualitative study. American journal of medical genetics. Part A. 2022 Feb:188(2):454-462. doi: 10.1002/ajmg.a.62534. Epub 2021 Oct 13 [PubMed PMID: 34643322]
Level 2 (mid-level) evidenceHunter AG, Bankier A, Rogers JG, Sillence D, Scott CI Jr. Medical complications of achondroplasia: a multicentre patient review. Journal of medical genetics. 1998 Sep:35(9):705-12 [PubMed PMID: 9733026]
Thompson NM, Hecht JT, Bohan TP, Kramer LA, Davidson K, Brandt ME, Fletcher JM. Neuroanatomic and neuropsychological outcome in school-age children with achondroplasia. American journal of medical genetics. 1999 Apr 16:88(2):145-53 [PubMed PMID: 10206234]
Fredwall SO, Steen U, de Vries O, Rustad CF, Eggesbø HB, Weedon-Fekjær H, Lidal IB, Savarirayan R, Månum G. High prevalence of symptomatic spinal stenosis in Norwegian adults with achondroplasia: a population-based study. Orphanet journal of rare diseases. 2020 May 25:15(1):123. doi: 10.1186/s13023-020-01397-6. Epub 2020 May 25 [PubMed PMID: 32450891]
Stender M, Pimenta JM, Cheung M, Irving M, Mukherjee S. Comprehensive literature review on the prevalence of comorbid conditions in patients with achondroplasia. Bone. 2022 Sep:162():116472. doi: 10.1016/j.bone.2022.116472. Epub 2022 Jun 18 [PubMed PMID: 35728791]
Kashanian A, Stadler JA 3rd, Danielpour M. Neurosurgical Evaluation and Management of Children with Achondroplasia. Neurosurgery clinics of North America. 2022 Jan:33(1):17-23. doi: 10.1016/j.nec.2021.09.003. Epub 2021 Oct 26 [PubMed PMID: 34801138]
Hecht JT, Butler IJ, Scott CI Jr. Long-term neurological sequelae in achondroplasia. European journal of pediatrics. 1984 Nov:143(1):58-60 [PubMed PMID: 6510432]
Leiva-Gea A, Martos Lirio MF, Barreda Bonis AC, Marín Del Barrio S, Heath KE, Marín Reina P, Guillén-Navarro E, Santos Simarro F, Riaño Galán I, Yeste Fernández D, Leiva-Gea I. Achondroplasia: Update on diagnosis, follow-up and treatment. Anales de pediatria. 2022 Dec:97(6):423.e1-423.e11. doi: 10.1016/j.anpede.2022.10.004. Epub 2022 Nov 5 [PubMed PMID: 36347803]
Silverman FN. Radiologic features of achondroplasia. Basic life sciences. 1988:48():31-44 [PubMed PMID: 3240266]
Cheung MS, Irving M, Cocca A, Santos R, Shaunak M, Dougherty H, Siddiqui A, Gringras P, Thompson D. Achondroplasia Foramen Magnum Score: screening infants for stenosis. Archives of disease in childhood. 2021 Feb:106(2):180-184. doi: 10.1136/archdischild-2020-319625. Epub 2020 Sep 3 [PubMed PMID: 32883660]
Jenko N, Connolly DJA, Raghavan A, Fernandes JA, Ushewokunze S, Elphick HE, Arundel P, Alhun U, Offiah AC. The (extended) achondroplasia foramen magnum score has good observer reliability. Pediatric radiology. 2022 Jul:52(8):1512-1520. doi: 10.1007/s00247-022-05348-0. Epub 2022 Apr 9 [PubMed PMID: 35396670]
Irving M, AlSayed M, Arundel P, Baujat G, Ben-Omran T, Boero S, Cormier-Daire V, Fredwall S, Guillen-Navarro E, Hoyer-Kuhn H, Kunkel P, Lampe C, Maghnie M, Mohnike K, Mortier G, Sousa SB. European Achondroplasia Forum guiding principles for the detection and management of foramen magnum stenosis. Orphanet journal of rare diseases. 2023 Jul 27:18(1):219. doi: 10.1186/s13023-023-02795-2. Epub 2023 Jul 27 [PubMed PMID: 37501185]
Cabet S, Szathmari A, Mottolese C, Franco P, Guibaud L, Rossi M, Di Rocco F. New insights in craniovertebral junction MR changes leading to stenosis in children with achondroplasia. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2022 Jun:38(6):1137-1145. doi: 10.1007/s00381-022-05514-7. Epub 2022 May 3 [PubMed PMID: 35505148]
Savarirayan R, Ireland P, Irving M, Thompson D, Alves I, Baratela WAR, Betts J, Bober MB, Boero S, Briddell J, Campbell J, Campeau PM, Carl-Innig P, Cheung MS, Cobourne M, Cormier-Daire V, Deladure-Molla M, Del Pino M, Elphick H, Fano V, Fauroux B, Gibbins J, Groves ML, Hagenäs L, Hannon T, Hoover-Fong J, Kaisermann M, Leiva-Gea A, Llerena J, Mackenzie W, Martin K, Mazzoleni F, McDonnell S, Meazzini MC, Milerad J, Mohnike K, Mortier GR, Offiah A, Ozono K, Phillips JA 3rd, Powell S, Prasad Y, Raggio C, Rosselli P, Rossiter J, Selicorni A, Sessa M, Theroux M, Thomas M, Trespedi L, Tunkel D, Wallis C, Wright M, Yasui N, Fredwall SO. International Consensus Statement on the diagnosis, multidisciplinary management and lifelong care of individuals with achondroplasia. Nature reviews. Endocrinology. 2022 Mar:18(3):173-189. doi: 10.1038/s41574-021-00595-x. Epub 2021 Nov 26 [PubMed PMID: 34837063]
Level 3 (low-level) evidenceKim J, Patel VJ, El Ahmadieh TY, Olson DM, Swift DM. Hydrocephalus in achondroplasia: efficacy of endoscopic third ventriculostomy. Journal of neurosurgery. Pediatrics. 2022 Mar 1:29(3):268-275. doi: 10.3171/2021.9.PEDS21242. Epub 2021 Dec 17 [PubMed PMID: 34920430]
Shoda K, Ohe N, Sasai H, Miyai M, Ohnishi H, Iwama T. Endoscopic third ventriculostomy for hydrocephalus in a patient with achondroplasia: a case report and literature review. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2021 Dec:37(12):3907-3911. doi: 10.1007/s00381-021-05129-4. Epub 2021 Mar 29 [PubMed PMID: 33779806]
Level 3 (low-level) evidenceSteinbok P, Hall J, Flodmark O. Hydrocephalus in achondroplasia: the possible role of intracranial venous hypertension. Journal of neurosurgery. 1989 Jul:71(1):42-8 [PubMed PMID: 2786928]
Level 3 (low-level) evidenceAkinnusotu O, Isaacs AM, Stone M, Bonfield CM. Neurosurgical management of cervicomedullary compression, spinal stenosis, and hydrocephalus in pediatric achondroplasia: a systematic review. Journal of neurosurgery. Pediatrics. 2023 Nov 1:32(5):597-606. doi: 10.3171/2023.6.PEDS23162. Epub 2023 Aug 18 [PubMed PMID: 37728398]
Level 1 (high-level) evidenceSandvik U, Ringvall E, Klangemo K, Hallgrimsdottir S, Gkourogianni A, Ottosson L, Svoboda J, Nilsson O. Management and outcomes of foramen magnum stenosis in children with achondroplasia at a single center over 15 years. Journal of neurosurgery. Pediatrics. 2024 Nov 1:34(5):470-478. doi: 10.3171/2024.6.PEDS23586. Epub 2024 Aug 30 [PubMed PMID: 39213664]
Fauroux B, AlSayed M, Ben-Omran T, Boero S, Boon M, Cormier-Daire V, Fredwall S, Guillen-Navarro E, Irving M, Kunkel P, Madureira N, Maghnie M, Milerad J, Mohnike K, Mortier G, Nobili L, Pejin Z, Sessa M, Sousa SB. Management of sleep-disordered breathing in achondroplasia: guiding principles of the European Achondroplasia Forum. Orphanet journal of rare diseases. 2025 May 15:20(1):233. doi: 10.1186/s13023-025-03717-0. Epub 2025 May 15 [PubMed PMID: 40375310]
Engberts AC, Jacobs WC, Castelijns SJ, Castelein RM, Vleggeert-Lankamp CL. The prevalence of thoracolumbar kyphosis in achondroplasia: a systematic review. Journal of children's orthopaedics. 2012 Mar:6(1):69-73. doi: 10.1007/s11832-011-0378-7. Epub 2011 Dec 3 [PubMed PMID: 22442656]
Level 1 (high-level) evidenceOmara C, Pieters L, Castelein RM, Sakkers RJB, Vleggeert-Lankamp CLA. Early evaluation and treatment of thoracolumbar kyphosis in children with achondroplasia. European spine journal : official publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research Society. 2025 Apr:34(4):1221-1228. doi: 10.1007/s00586-025-08692-5. Epub 2025 Feb 3 [PubMed PMID: 39894831]
Shirley ED, Ain MC. Achondroplasia: manifestations and treatment. The Journal of the American Academy of Orthopaedic Surgeons. 2009 Apr:17(4):231-41 [PubMed PMID: 19307672]
Thomeer RT, van Dijk JM. Surgical treatment of lumbar stenosis in achondroplasia. Journal of neurosurgery. 2002 Apr:96(3 Suppl):292-7 [PubMed PMID: 11990837]
Carlisle ES, Ting BL, Abdullah MA, Skolasky RL, Schkrohowsky JG, Yost MT, Rigamonti D, Ain MC. Laminectomy in patients with achondroplasia: the impact of time to surgery on long-term function. Spine. 2011 May 15:36(11):886-92. doi: 10.1097/BRS.0b013e3181e7cb2a. Epub [PubMed PMID: 20739914]
Hariharan AR, Nugraha HK, Huser AJ, Feldman DS. Surgery for Spinal Stenosis in Achondroplasia: Causes of Reoperation and Reduction of Risks. Journal of pediatric orthopedics. 2024 Aug 1:44(7):448-455. doi: 10.1097/BPO.0000000000002687. Epub 2024 Apr 9 [PubMed PMID: 38595075]
Abu Al-Rub Z, Lineham B, Hashim Z, Stephenson J, Arnold L, Campbell J, Loughenbury P, Khan A. Surgical treatment of spinal stenosis in achondroplasia: Literature review comparing results in adults and paediatrics. Journal of clinical orthopaedics and trauma. 2021 Dec:23():101672. doi: 10.1016/j.jcot.2021.101672. Epub 2021 Oct 30 [PubMed PMID: 34790562]
Fredwall SO, Linge J, Leinhard OD, Kjønigsen L, Eggesbø HB, Weedon-Fekjær H, Lidal IB, Månum G, Savarirayan R, Tonstad S. Cardiovascular risk factors and body composition in adults with achondroplasia. Genetics in medicine : official journal of the American College of Medical Genetics. 2021 Apr:23(4):732-739. doi: 10.1038/s41436-020-01024-6. Epub 2020 Nov 18 [PubMed PMID: 33204020]
Saint-Laurent C, Garde-Etayo L, Gouze E. Obesity in achondroplasia patients: from evidence to medical monitoring. Orphanet journal of rare diseases. 2019 Nov 14:14(1):253. doi: 10.1186/s13023-019-1247-6. Epub 2019 Nov 14 [PubMed PMID: 31727132]
Savarirayan R, Tofts L, Irving M, Wilcox W, Bacino CA, Hoover-Fong J, Ullot Font R, Harmatz P, Rutsch F, Bober MB, Polgreen LE, Ginebreda I, Mohnike K, Charrow J, Hoernschemeyer D, Ozono K, Alanay Y, Arundel P, Kagami S, Yasui N, White KK, Saal HM, Leiva-Gea A, Luna-González F, Mochizuki H, Basel D, Porco DM, Jayaram K, Fisheleva E, Huntsman-Labed A, Day J. Once-daily, subcutaneous vosoritide therapy in children with achondroplasia: a randomised, double-blind, phase 3, placebo-controlled, multicentre trial. Lancet (London, England). 2020 Sep 5:396(10252):684-692. doi: 10.1016/S0140-6736(20)31541-5. Epub [PubMed PMID: 32891212]
Level 1 (high-level) evidenceSavarirayan R, Irving M, Bacino CA, Bostwick B, Charrow J, Cormier-Daire V, Le Quan Sang KH, Dickson P, Harmatz P, Phillips J, Owen N, Cherukuri A, Jayaram K, Jeha GS, Larimore K, Chan ML, Huntsman Labed A, Day J, Hoover-Fong J. C-Type Natriuretic Peptide Analogue Therapy in Children with Achondroplasia. The New England journal of medicine. 2019 Jul 4:381(1):25-35. doi: 10.1056/NEJMoa1813446. Epub 2019 Jun 18 [PubMed PMID: 31269546]
Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocrine reviews. 2000 Feb:21(1):23-39 [PubMed PMID: 10696568]
Krakow D, Rimoin DL. The skeletal dysplasias. Genetics in medicine : official journal of the American College of Medical Genetics. 2010 Jun:12(6):327-41. doi: 10.1097/GIM.0b013e3181daae9b. Epub [PubMed PMID: 20556869]
Almeida MR, Campos-Xavier AB, Medeira A, Cordeiro I, Sousa AB, Lima M, Soares G, Rocha M, Saraiva J, Ramos L, Sousa S, Marcelino JP, Correia A, Santos HG. Clinical and molecular diagnosis of the skeletal dysplasias associated with mutations in the gene encoding Fibroblast Growth Factor Receptor 3 (FGFR3) in Portugal. Clinical genetics. 2009 Feb:75(2):150-6. doi: 10.1111/j.1399-0004.2008.01123.x. Epub [PubMed PMID: 19215249]
Level 3 (low-level) evidenceAdam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, French T, Savarirayan R. Thanatophoric Dysplasia. GeneReviews(®). 1993:(): [PubMed PMID: 20301540]
Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, Israel J, Rosengren SS, Webster MK, Donoghue DJ, Francomano CA. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. American journal of human genetics. 2000 Dec:67(6):1411-21 [PubMed PMID: 11055896]
Level 3 (low-level) evidenceTavormina PL, Bellus GA, Webster MK, Bamshad MJ, Fraley AE, McIntosh I, Szabo J, Jiang W, Jabs EW, Wilcox WR, Wasmuth JJ, Donoghue DJ, Thompson LM, Francomano CA. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. American journal of human genetics. 1999 Mar:64(3):722-31 [PubMed PMID: 10053006]
Zankl A, Elakis G, Susman RD, Inglis G, Gardener G, Buckley MF, Roscioli T. Prenatal and postnatal presentation of severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) due to the FGFR3 Lys650Met mutation. American journal of medical genetics. Part A. 2008 Jan 15:146A(2):212-8 [PubMed PMID: 18076102]
Dai L, Xie L, Wang Y, Mao M, Li N, Zhu J, Kim C, Zhang Y. A novel COMP mutation in a pseudoachondroplasia family of Chinese origin. BMC medical genetics. 2011 May 21:12():72. doi: 10.1186/1471-2350-12-72. Epub 2011 May 21 [PubMed PMID: 21599986]
Level 3 (low-level) evidenceHecht JT, Francomano CA, Horton WA, Annegers JF. Mortality in achondroplasia. American journal of human genetics. 1987 Sep:41(3):454-64 [PubMed PMID: 3631079]