Introduction
Double-outlet right ventricle is a highly complex and heterogeneous group of congenital heart diseases with an anatomic spectrum ranging from tetralogy of Fallot to transposition of the great arteries. First described in 1957, double outlet right ventricle represents 1% to 3% of patients with congenital heart defects and 8% to 9% of all conotruncal anomalies.[1][2] The physiology and the most appropriate surgical treatment require a firm understanding of the relationship between the great vessels and the location of the ventricular septal defect. The spiraling septation of the primitive truncus determines the ventricular septal defect location. Many classification schemes have been proposed during the past 50 years and continue to be debated.[3][4] Surgical treatment of double outlet right ventricle continues to evolve, and techniques range widely from biventricular repairs with an intraventricular baffle to univentricular staged palliation.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of congenital heart disease (CHD) is multifactorial, with increasingly recognized genetic causes. DORV is a conotruncal defect that results from abnormalities in the second heart field and neural crest during embryonic development. Epigenetic and environmental factors have been implicated in the development of DORV, including DNA methylation, teratogen exposure, and insufficient maternal folate consumption.[5] More than 40% of patients with DORV have chromosomal abnormalities.[6]
Trisomy 21 occurs in 4% of patients with DORV; CHD occurs in 90% of patients with trisomy 13, and DORV occurs in 11% of patients with trisomy 13 and CHD; CHD occurs in 85% of patients with trisomy 18, and DORV occurs in 13% of patients with trisomy 18 and CHD.[2][7] In addition, 10% of patients with DORV have chromosome 8 abnormalities, and 7% have 22q11.2 deletion syndrome. Other associated genetic syndromes include 5p15.2 deletion, also known as cri du chat syndrome, and 11q terminal deletion, also known as Jacobsen syndrome.[8]
Mutations in the T-box transcription factor 5 gene (TBX5) have been implicated in Holt-Oram syndrome, and 22q11.2 deletion has been implicated in DiGeorge syndrome. Familial recurrence of DORV has been reported in 5% of cases, and DORV has also been associated with parental consanguinity. A table of single-gene defects seen in patients with DORV can be found in the article by Cornelia Dorn et al and includes BCL6 corepressor gene (BCOR), ISL LIM homeobox 1 gene (ISL1), NK2 homeobox 5 gene (NKX2-5), NK2 homeobox 6 gene (NKX2-6), paired-like homeodomain transcription factor 2 gene (PITX2), zinc finger protein, FOG family member 2 gene (ZFPM2), Zic family member 3 gene (ZIC3), cripto, FRL-1, cryptic family 1 gene (CFC1), growth differentiation factor 1 gene (GDF1), and nodal growth differentiation factor gene (NODAL), among others.[6]
Epidemiology
DORV occurs at rates of 3 to 9 per 100,000 live births.[8] This heterogeneous group of congenital heart defects represents 1% to 3% of patients with congenital heart disease and 8% to 9% of all conotruncal anomalies.[1][2] DORV is more common in male patients, with a ratio of 2.68:1.[9] Overall, the birth prevalence of congenital heart disease has been rising, with an increase seen from 4.5 per 1000 live births in 1970 to 1974 to 9.4 per 1000 live births in 2010 to 2017. Geographical variations have also been observed, with the birth prevalence of congenital heart disease in Africa being 2.1 per 1000 live births, approximately one-quarter of the rates in Asia, Europe, and North and South America.[10]
Pathophysiology
DORV presents with a wide spectrum of anatomical variants and subsequent pathophysiology.[8] One suggested mechanism of heart failure in DORV is that chronic arterial hypoxemia triggers hypoxia-inducible factor activation, leading to secondary complications such as excessive red blood cell production, coagulation abnormalities, and pathologic angiogenesis. Furthermore, hemodynamic stress causes myocardial remodeling, characterized by a return to the fetal gene program, cellular calcium overload, and progressive myocardial fibrosis leading to heart failure.[11][12]
Anatomy
Any discussion of DORV anatomy should start with the Congenital Heart Surgery Nomenclature and Database Project.[3] The project defines DORV, as Anderson and colleagues did in 1975, as “a type of ventriculo-arterial connection in which both great vessels arise entirely or predominantly from the right ventricle.” Importantly, this lesion is set apart from other lesions because the ventricular septal defect (VSD) forms “an integral part of the left ventricular outflow tract.”[13] Accurate identification of this anatomy is critical when considering surgical repair because the result must create an unobstructed left ventricular outflow tract. Many have argued that the classification of these lesions is less important than a precise, detailed anatomical delineation to guide surgical decision-making.[14][15]
Four Subtypes
The most widely used classification of DORV was published in 1972 by Lev et al.[16] This classification included the following 4 subtypes by VSD location:
1. Subaortic ventral septal defect: A subaortic VSD is the most common subtype of DORV, occurring in 50% of patients.[17] In this subtype, left ventricular blood flow is directed into the aorta. The severity of pulmonary stenosis determines the physiology in these patients.
When pulmonary stenosis or right ventricular outflow tract obstruction is present, the physiology is similar to that of tetralogy of Fallot with pulmonary stenosis. Patients can be hemodynamically balanced if pulmonary stenosis prevents pulmonary overcirculation or cyanotic if the pulmonary stenosis is severe, resulting in insufficient pulmonary blood flow and right-to-left shunting. When pulmonary stenosis is absent, the physiology is similar to that of a large VSD with pulmonary overcirculation, high oxygen saturations, and heart failure.
2. Subpulmonary ventricular septal defect (Taussig-Bing anomaly): A subpulmonary VSD is the second most common type of DORV, occurring in 30% of patients.[17] The physiology in these patients is similar to that of patients with dextrotransposition of the great arteries: left ventricular blood flows across the VSD into the pulmonary artery, and right ventricular blood flows into the aorta. When there is no obstruction to blood flow into the pulmonary artery, pulmonary overcirculation occurs. Cyanosis also occurs when right ventricular blood flows into the aorta, with the degree of cyanosis depending on the degree of mixing via an atrial septal defect, VSD, or patent ductus arteriosus.
Although sometimes used interchangeably, the term Taussig-Bing refers to a special type of DORV with subpulmonary VSD. In addition to a subpulmonary VSD, the great arteries are side by side, with the aorta to the right and the pulmonary artery to the left. Patients with Taussig-Bing anomaly have a high rate (70%) of concomitant aortic arch obstruction, including coarctation.[18] Coronary anomalies and subaortic stenosis are also frequently seen. In addition to arch obstruction or coarctation, total anomalous pulmonary venous return and complete atrioventricular canal are also associated.
3. Doubly committed (or subarterial) ventricular septal defect: In a doubly committed VSD subtype, the VSD lies directly beneath both the aorta and the pulmonary artery. Thus, blood flow across the VSD is directed into both great vessels. Fibrous continuity is seen between the arterial valves. The physiology of this subtype is equivalent to that of DORV with a subaortic VSD, with or without pulmonary stenosis. When pulmonary stenosis is present, its severity determines the physiology.
4. Noncommitted ventricular septal defect: In a noncommitted VSD subtype, the VSD is remote from both the aorta and pulmonary artery, and blood flow is not directed across either outflow tract. The VSD is typically an inlet type or apical VSD. The physiology of this subtype is similar to that of DORV with subaortic VSD, with or without pulmonary stenosis. When pulmonary stenosis is present, its severity determines the physiology.
Although useful for considering surgical interventions, this classification scheme has fallen short for several reasons. The physiology of DORV depends not only on the VSD location but also on the relationship of the great vessels, and the location of the VSD is independent of the great artery relationship. Additionally, DORV can rarely be associated with discordant or univentricular atrioventricular connections, atrioventricular valve atresia, or atrial isomerism.[3]
The anatomical and hemodynamic classification: Introduced in 2000, this widely accepted system classifies DORV based on anatomy, clinical presentation, and blood flow patterns. The system divides DORV into 4 main types. The VSD type has a subaortic or subarterial defect, so blood from the left ventricle reaches the aorta more directly; these patients are usually not cyanotic, but they often develop pulmonary overcirculation and pulmonary hypertension.
The tetralogy type also has a subaortic or subarterial defect, but with pulmonary stenosis, which reduces pulmonary blood flow and leads to cyanosis. The transposition of the great arteries type is characterized by a subpulmonary defect, causing severe cyanosis because oxygenated blood is directed mainly to the pulmonary artery while deoxygenated blood enters the aorta; this type is also associated with pulmonary overcirculation and pulmonary hypertension. The noncommitted type may occur with or without pulmonary stenosis, and the clinical presentation can be quite variable, depending on the proximity of the defect to the arterial valves and on the presence of pulmonary stenosis.[8][19] In the Congenital Heart Surgery Nomenclature and Database Project, DORV with discordant atrioventricular connections is grouped with corrected (discordant) transposition of the great arteries (and related malformations), while DORV with univentricular atrioventricular connections, atrioventricular valve atresia, or atrial isomerism is grouped with other single ventricle lesions.[3]
Natural History
The natural history of DORV is quite variable. The relationship of the VSD to the great vessels, the presence and degree of pulmonary stenosis, and any additional cardiac defects dictate the timing and type of clinical presentation. The physiology of DORV must be understood within the context of the VSD location, the relationship of the great vessels, and whether any outflow tract obstruction exists. The location of the VSD is independent of the great artery relationship. When the VSD is committed or directed into a particular outflow tract, it can be subaortic, subpulmonary, or doubly committed. The types of presentation are as heterogeneous as the spectrum of underlying anatomy.
In general, patients with DORV present early in life, with a mean age of 2 months. One category of presentation is congestive heart failure and failure to thrive, such as in patients without pulmonary stenosis with unrestricted subaortic, doubly committed, or noncommitted VSDs or patients with left ventricular outflow tract obstruction leading to pulmonary venous obstruction. These patients often present with pulmonary overcirculation that can require aggressive diuresis, respiratory support, and even intubation.[8] Without repair, these patients can develop chronic pulmonary vascular obstructive disease by 1 year of age.
Conversely, when pulmonary blood flow is inadequate, patients can present with cyanosis, such as patients with restricted pulmonary blood flow or streaming of highly saturated left ventricular blood to the pulmonary artery, as occurs in Taussig-Bing anomaly. The physiology is similar to that of dextro-transposition of the great arteries. These patients may require prostaglandin E1 infusion or atrial septostomy immediately after birth to promote adequate mixing. A subset of patients may initially have sufficient pulmonary blood flow but gradually develop progressive cyanosis over time. Lastly, patients with DORV and left ventricular outflow tract obstruction can present in cardiogenic shock.
History and Physical
The patient's history and clinical presentation depend on the DORV subtype's physiology. In general, patients with DORV usually present in the neonatal period, often after receiving an antenatal diagnosis. Results from a recent study showed that the mean gestational age at first diagnosis was approximately 21 weeks.[17] The clinical presentation depends on the patient's specific anatomic features, with the location of the VSD and degree of outflow tract obstruction dictating the continuum between pulmonary overcirculation and insufficient pulmonary blood flow with cyanosis. When left ventricular outflow tract obstruction or aortic arch obstruction is present, the patient may present in circulatory collapse.
For patients presenting with pulmonary overcirculation, such as those with a subaortic VSD and no pulmonary stenosis, the constellation of symptoms is consistent with heart failure and can include tachypnea, dyspnea, diaphoresis, feeding intolerance, and failure to thrive. Hepatomegaly may also be seen on physical examination. A holosystolic murmur is present and best heard at the left sternal border. For patients with pulmonary stenosis, a systolic ejection murmur may also be heard, and when pulmonary stenosis is severe, patients present with cyanosis due to decreased pulmonary blood flow.
Evaluation
Diagnostic studies to evaluate patients with DORV should include chest radiography, echocardiography, and, when indicated, cross-sectional imaging. CT, MRI, and cardiac catheterization can be considered in selected patients. Additional imaging selection depends on the suspected anatomy, surgical planning needs, and hemodynamic questions that remain after echocardiography.
Chest radiography findings depend on the DORV subtype. For example, in patients with subaortic VSD without pulmonary stenosis or right ventricular outflow tract obstruction, cardiomegaly and plethoric lungs are seen, consistent with heart failure. However, in patients with pulmonary stenosis, chest radiography findings show reduced pulmonary vascular markings.
Transthoracic echocardiography is the mainstay of evaluation and the gold standard for confirming the diagnosis of DORV. Clinicians should use a systematic approach, looking at atrial and ventricular morphology, systemic and pulmonary venous return, interatrial and interventricular communications, atrioventricular valve anatomy and function, the size and relationship of the great arteries, aortic and pulmonary valves, aortic arch, coronary artery anatomy, outflow tracts, and degree of obstruction. Features of particular importance in DORV include the following: 1) atrioventricular valves, including assessment for a common atrioventricular valve or straddling mitral valve, which would complicate closure of a subpulmonary VSD; 2) the distance between the tricuspid valve and pulmonary valve; and 3) VSD location, size, and number.[20]
CT can be helpful in evaluating complex intracardiac anatomy, especially when paired with 3D modeling and 3D printing, if available; this information can be critical for VSD closure and baffle planning. When the distance between the tricuspid and pulmonary valves is small, the patient has a risk of baffle obstruction. CT can also be used to evaluate the relationship between the ventricular chambers and the aorta and pulmonary artery, and to further characterize the VSD. Coronary artery anatomy can also be further delineated, which is helpful if an arterial switch operation is required or if the right ventricular outflow tract requires augmentation.[21][22]
MRI can be used in selected patients to measure pulmonary vascular resistance or to measure pulmonary blood flow relative to systemic blood flow (Qp:Qs). MRI can also be used to delineate coronary anatomy. Cardiac magnetic resonance offers an important advantage over echocardiography by consistently quantifying cardiac function. Cardiac magnetic resonance can reliably measure right and left ventricular volumes at end diastole and end systole, ejection fraction, stroke volume, pulmonary-to-systemic flow ratio, and the severity of regurgitation using cine sequences and phase-contrast imaging. In patients with DORV, this information is especially useful because treatment is determined not only by anatomy but also by whether both ventricles are adequate and whether the circulation can support biventricular repair.[23]
Cardiac catheterization can also be used to measure pulmonary vascular resistance and Qp:Qs ratio or to delineate coronary anatomy. In addition to diagnostic information, certain subtypes of DORV may necessitate establishing a stable source of pulmonary blood flow, which can be done in the catheterization laboratory. For example, in patients with severe pulmonary stenosis, patent ductus arteriosus stents have been placed to maintain ductal-dependent pulmonary blood flow. Right ventricular outflow tract stents are used less commonly.
Treatment / Management
Unless the patient is completely balanced, an earlier intervention is often necessary before complete surgical repair. Depending on the degree of pulmonary stenosis, intervention includes either restricting or enhancing pulmonary blood flow. If a subsequent biventricular repair will not be feasible, the pulmonary vascular bed must be protected until the first of the 2 staged cavopulmonary connections can occur. Please see StatPearls' companion reference, "Fontan Completion," for further information.
For patients without pulmonary stenosis and large VSDs, pulmonary overcirculation and increasing heart failure symptoms occur as the pulmonary vascular resistance falls. Pulmonary blood flow can be restricted in these children by surgical placement of a pulmonary artery band. Pulmonary flow restrictors placed in the catheterization laboratory have also been used to restrict pulmonary blood flow before complete surgical repair.[24](B3)
For patients with significant pulmonary stenosis or pulmonary atresia, inadequate pulmonary blood flow may necessitate establishing a durable source of pulmonary blood flow. This source can be established either surgically or in the catheterization laboratory and includes placement of a right ventricle–to–pulmonary artery shunt, modified Blalock-Taussig-Thomas shunt, patent ductus arteriosus stent, or right ventricular outflow tract stent. Right ventricular outflow tract stents are less common and carry the risk of crossing the pulmonary valve, which may necessitate later transannular patch and subsequent pulmonary valve replacement. Thus, right ventricular outflow tract stents are only used in cases where the pulmonary valve is unlikely to be spared. Surgical intervention for complete repair is guided by the underlying anatomy and physiology, ranging from single ventricle palliation to biventricular surgical repair, either primary or staged.
Subaortic Ventricular Septal Defect or Doubly Committed Ventricular Septal Defect
In the most common type of DORV with subaortic VSD, when pulmonary stenosis is absent, complete surgical repair involves closing the VSD with a left ventricle-to-aorta baffle (see Image. Surgical Anatomy of Double Outlet Right Ventricle), and (see Image. Surgical Repair of Double Outlet Right Ventricle). This repair directs left ventricular blood out through the aorta and right ventricular blood to the pulmonary artery. If the VSD is not large enough to allow unrestricted flow into the aorta, the defect requires enlargement. The same repair strategy is used for DORV with doubly committed VSD. If pulmonary stenosis or atresia is present, the operation is similar to that of a tetralogy of Fallot repair, with potential for pulmonary valvuloplasty, augmentation of the right ventricular outflow tract, right ventricular muscle bundle resection, transannular patch versus valve-sparing repair, or valved conduit.
Subpulmonary Ventricular Septal Defect
Surgical repair for a subpulmonary VSD depends on the great artery anatomy. When no outflow tract obstruction is present, as in transposition of the great arteries, an arterial switch procedure (Jatene) with the LeCompte maneuver and closure of the VSD to the pulmonary artery is performed. When the great arteries are side by side, such as in Taussig-Bing anomaly, an arterial switch operation may still be possible.
When arch hypoplasia is also present, concomitant arch repair must also be performed. When an arterial switch operation is not possible, such as in the presence of concomitant subpulmonary stenosis, several surgical options can reconstruct the right ventricular outflow tract. In a Rastelli operation, an intraventricular repair with tunneling of the aorta to the left ventricle through the VSD and right ventricle–to–pulmonary artery conduit placement is performed. In a réparation à l’étage ventriculaire procedure, the aorta is tunneled to the left ventricle through the VSD, with a LeCompte procedure and direct anastomosis of the pulmonary trunk bifurcation to the right ventriculotomy, with patch augmentation. When other associated congenital anomalies are present, concurrent repair may be required, such as for total anomalous pulmonary venous return or complete atrioventricular canal.
Noncommitted Ventricular Septal Defect
In DORV, when the distance between the tricuspid valve and pulmonary valve is too short, the baffle or pulmonary valve can become obstructed. In these patients, a single ventricle pathway may be necessary. When a single-ventricle pathway is needed, the pulmonary vascular bed must be protected until the first of the 2 staged cavopulmonary connections can be performed. This protection may require placement of a pulmonary artery band or bilateral pulmonary flow restrictors.
Straddling the Mitral Valve
A straddling mitral valve complicates VSD closure in DORV. Repositioning of the straddling mitral valve chordal attachments to the baffle or to the crest of the septum may be considered in patients with this anatomy. When the tricuspid or mitral valve apparatus is complex, or when multiple complex VSDs are present, septation may not be feasible, and a single-ventricle pathway may be necessary. Early intervention to protect the pulmonary vascular bed with a pulmonary artery band or bilateral pulmonary flow restrictors allows growth and future reevaluation of the ability to septate.
Differential Diagnosis
Differential diagnoses include ventricular septal defect, tetralogy of Fallot, pulmonary atresia or stenosis, and transposition of the great arteries in patients with DORV and subpulmonary ventricular septal defect.
Pertinent Studies and Ongoing Trials
The largest retrospective study of this rare congenital lesion examined 1135 patients at a single center, Fuwai Hospital in Beijing, China, between 2001 and 2023. Results from the study showed that with detailed anatomic assessment and an appropriately chosen surgical strategy, most children with DORV can achieve excellent long-term survival. However, patients with transposition of the great arteries–type anatomy or who require palliative surgical procedures face a higher risk and may require closer follow-up and more intensive care optimization.[25]
Prognosis
In the current era, surgical results for biventricular repair are good. Results from 1 recent review showed early surgical mortality in 4.6% of patients and late mortality in 3.4% of patients. Overall survival was 91.9%, 90.8%, 89.5%, and 89.5% at 1, 2, 5, and 10 years, respectively. Freedom from surgical reintervention was 88.0%, 79.0%, 74.9%, and 69.4% at 1, 2, 5, and 10 years, respectively.[26]
Complications
Historically, surgical treatment of DORV was marked by postoperative complications and a high risk of sudden death.[27] Early mortality was 15% in this patient cohort due to low cardiac output syndrome, high residual right ventricular pressure, anomalous coronary artery injury, infection, and hemorrhage. In this cohort, most late deaths were due to arrhythmia. Results from another retrospective review from a single center found a perioperative mortality of 19%, and of the patients who survived to hospital discharge, 25% subsequently died.[28] Most of these deaths were sudden deaths attributed to cardiac causes, including fibrosis of the conduction system, ruptured septal aneurysm, surgical atrioventricular block, and ventricular or supraventricular tachyarrhythmias.
More recently, postoperative complications are similar in nature to those of other pediatric congenital cardiac surgical procedures.[29] Immediately upon weaning from cardiopulmonary bypass, patients may exhibit atrial or ventricular arrhythmias, which may require temporary pacing. Postoperative echocardiography findings may demonstrate the presence of ventricular dysfunction, residual intracardiac shunt, ventricular outflow tract obstruction, or valvular dysfunction.
Other immediate postoperative problems may include bleeding, myocardial edema, and pulmonary compliance problems. Low cardiac output syndrome may be present and predispose patients to multiple organ dysfunction syndrome. Hypertension due to increased systemic vascular resistance may increase bleeding at suture sites and impair ventricular performance. Junctional ectopic tachycardia is common and may affect cardiac output. Pacing, cooling, amiodarone, and dexmedetomidine may all be useful in treating junctional ectopic tachycardia.
Results from a recent retrospective cohort study showed midterm outcomes of patients with DORV treated with biventricular repair.[30] In 238 patients with a median follow-up of 20 months, cardiac transplant–free survival was 90% at 1 year and 89% at 3 years. Freedom from surgical reintervention, as mentioned above, was lower at 1 year (82%) and 3 years (65%). Reoperations were performed for left ventricular outflow tract obstruction, right ventricular outflow tract obstruction, valve repair, residual ventricular septal defect, ventricular septal defect enlargement, and pacemaker insertion for complete heart block. Due to the risks of long-term complications, patients should receive lifelong close monitoring from an expert congenital heart disease team.
Postoperative and Rehabilitation Care
Postoperative care following DORV repair should adhere to current standards of care in a dedicated cardiac intensive care unit with interdisciplinary management. Continuous monitoring with electrocardiography, invasive arterial pressure, central venous pressure or right atrial pressure, and near-infrared spectroscopy is routine. Temporary pacing wires can be used to augment the patient's heart rate and cardiac output to treat bradycardia, heart block, or junctional ectopic tachycardia.
Chest tubes are placed to monitor postoperative bleeding. Common postoperative treatment strategies for children with preoperative left-to-right shunts include milrinone for inotropy, lusitropy, and afterload reduction. Patients should be weaned from respiratory support as tolerated. As with surgical treatment, postoperative care depends largely on the type of repair, whether initial palliation or definitive repair was performed, and whether the patient follows a single-ventricle or biventricular pathway.
Deterrence and Patient Education
Perioperative patient and family education should focus on recognizing complications and reporting them to the care team. Education may include recognition of respiratory distress, congestive heart failure, arrhythmia, and chest pain. The family should also know how to care for the surgical wound and recognize incision complications such as infection or fever.
Pearls and Other Issues
Pearls regarding DORV include:
- Double outlet right ventricle is a spectrum of congenital heart disease in which more than 50% of both great arteries arise from the morphologic right ventricle. The presentation and pathophysiology are determined by the relationship of the ventricular septal defect to the great arteries and by the presence of any outflow tract obstruction. These factors also determine treatment.
- When a subaortic VSD, the most common subtype, is present, the pathophysiology depends on the degree of pulmonary stenosis or right ventricular outflow tract obstruction that affects the pulmonary circulation. The spectrum of disease may present similarly to tetralogy of Fallot or with pulmonary overcirculation. Treatment may involve baffling the left ventricular outflow to the aorta. When pulmonary stenosis is present, the obstruction should be relieved. When the VSD is restrictive, the defect should be enlarged.
- When a doubly committed VSD is present, the defect sits directly between the pulmonary and aortic valves, and flow is directed to both great arteries. The presentation can be similar to that of subaortic VSD. Treatment similarly requires ensuring left ventricular outflow to the aorta and unobstructed right ventricular–to–pulmonary artery blood flow.
- A subpulmonary VSD, the second most common type, clinically presents similarly to dextrotransposition of the great arteries. Left ventricular blood flows across the VSD to the pulmonary arteries, and right ventricular blood flows to the aorta. Patients have cyanosis depending on the degree of blood mixing. When no outflow tract obstruction is present, treatment may involve the arterial switch operation with the LeCompte maneuver, plus VSD closure and baffling of the VSD to the pulmonary artery. A Taussig-Bing anomaly is defined by DORV with a subpulmonary VSD and side-by-side great arteries. Surgical treatment may involve an arterial switch operation.
- Noncommitted VSDs are remote from both the aorta and pulmonary artery, and the pathophysiology is similar to that of DORV with subaortic VSD, with or without pulmonary stenosis. Complex associated abnormalities may be present, and the dynamic physiology may require different initial treatment strategies. For example, when unobstructed pulmonary blood flow is present, treatment requires prevention of pulmonary overcirculation. Other patients may have inadequate pulmonary blood flow and require initial creation of pulmonary blood flow.
- Surgical outcomes are overall very good, with high rates of operative, midterm, and long-term survival. Reintervention may be required for ventricular outflow obstruction or other residual or recurrent problems. In DORV, precise identification of the VSD is critical for safe surgical repair. The true VSD is the opening used to construct the intraventricular tunnel from the left ventricle to the aorta, whereas the separate superior opening near the ventriculoinfundibular fold represents the left ventricular exit and should not be mistaken for the VSD. Accurate distinction between the true VSD and the left ventricular exit is essential because the left ventricle in DORV lacks a normal outlet portion, and inadvertent closure of its exit would be fatal.
Enhancing Healthcare Team Outcomes
An interdisciplinary conference that includes congenital cardiac surgeons, pediatric cardiologists, cardiac anesthesiologists, cardiac intensivists, imagers, nurses, advanced practitioners, and pharmacists is now the standard of care for preoperative decision-making and planning. Intraoperatively, close collaboration among the surgeon, cardiac anesthesiologist, perfusionist, and nursing team is essential. Collaboration between the surgeon and echocardiographer is particularly essential for determining the final operative plan after the preoperative transesophageal echocardiogram and for evaluating the repair. Postoperatively, daily interdisciplinary collaboration is essential for optimal outcomes.
A strategic approach is equally crucial, involving evidence-based strategies to optimize treatment plans and minimize adverse effects. Ethical considerations must guide decision-making, ensuring informed consent and respecting patient autonomy in treatment choices. Each healthcare professional must be aware of their responsibilities and contribute their unique expertise to the patient's care plan, fostering an interdisciplinary approach.
Effective interprofessional communication is paramount, allowing seamless information exchange and collaborative decision-making among team members. Care coordination plays a pivotal role in ensuring that the patient's journey from diagnosis to treatment and follow-up is well-treated, minimizing errors and enhancing patient safety. By embracing these principles of skill, strategy, ethics, responsibilities, interprofessional communication, and care coordination, healthcare professionals can deliver patient-centered care, ultimately improving patient outcomes and enhancing team performance in patients with double outlet right ventricle.
Media
(Click Image to Enlarge)
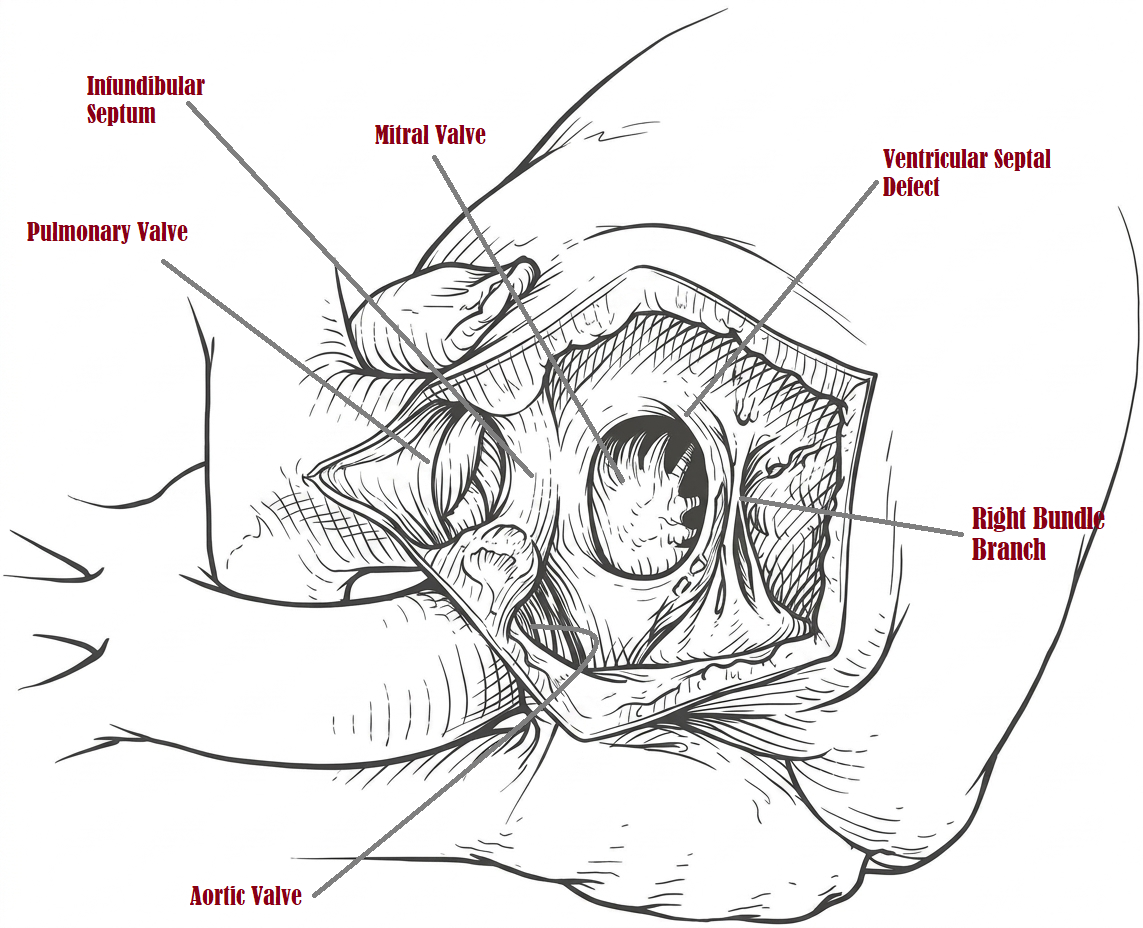
Surgical Anatomy of Double Outlet Right Ventricle. The origin of both the aorta and pulmonary artery from the right ventricle and their spatial relationship to a subaortic ventricular septal defect before the placement of a corrective intracardiac baffle.
Contributed by MH Mohammed Alahmadi, MBBS, MS, CHPE, MBA
(Click Image to Enlarge)
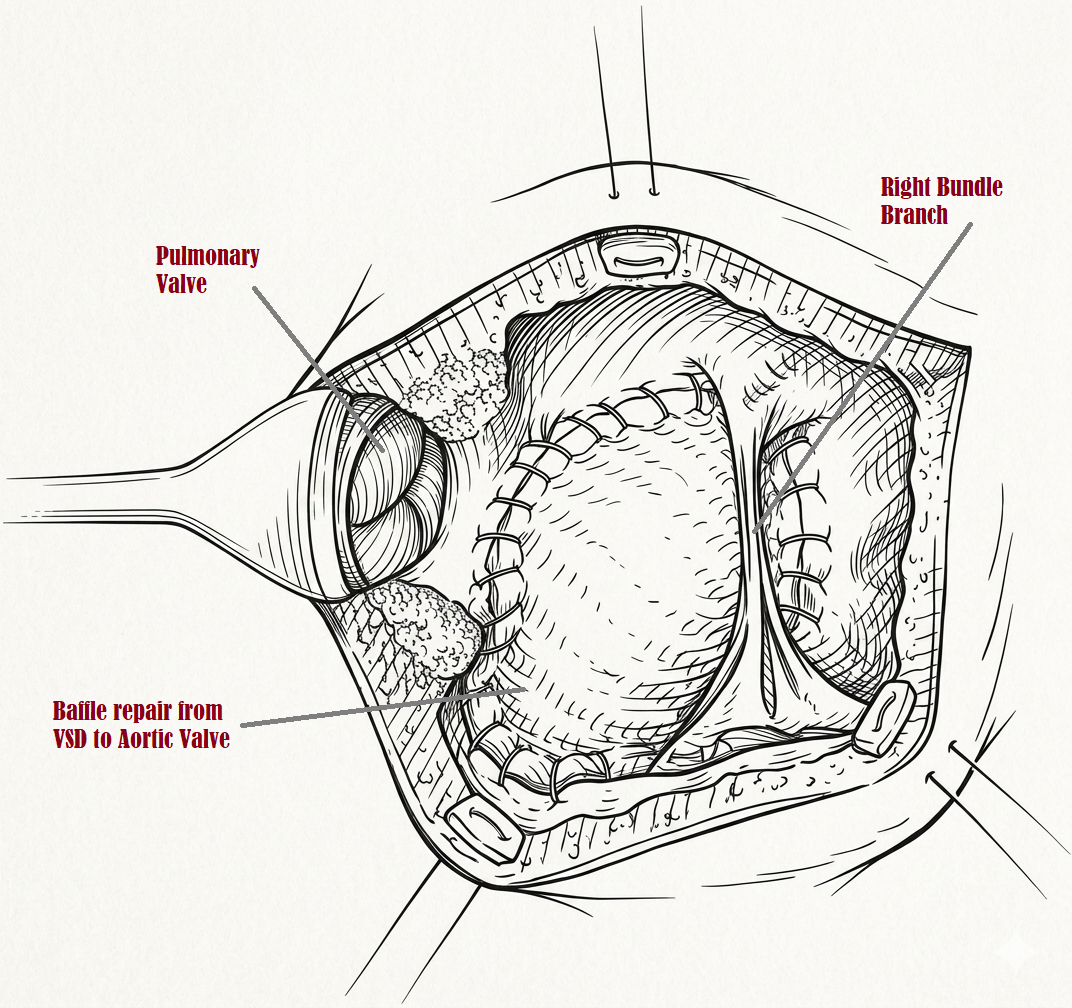
Surgical Repair of Double Outlet Right Ventricle. The completion of a biventricular repair, featuring an intracardiac baffle that routes flow from the ventricular septal defect to the aortic valve while maintaining patency of the pulmonary outflow tract.
Contributed by MH Mohammed Alahmadi, MBBS, MS, CHPE, MBA
References
WITHAM AC. Double outlet right ventricle; a partial transposition complex. American heart journal. 1957 Jun:53(6):928-39 [PubMed PMID: 13424473]
Obler D, Juraszek AL, Smoot LB, Natowicz MR. Double outlet right ventricle: aetiologies and associations. Journal of medical genetics. 2008 Aug:45(8):481-97. doi: 10.1136/jmg.2008.057984. Epub 2008 May 2 [PubMed PMID: 18456715]
Walters HL 3rd, Mavroudis C, Tchervenkov CI, Jacobs JP, Lacour-Gayet F, Jacobs ML. Congenital Heart Surgery Nomenclature and Database Project: double outlet right ventricle. The Annals of thoracic surgery. 2000 Apr:69(4 Suppl):S249-63 [PubMed PMID: 10798433]
Ebadi A, Spicer DE, Backer CL, Fricker FJ, Anderson RH. Double-outlet right ventricle revisited. The Journal of thoracic and cardiovascular surgery. 2017 Aug:154(2):598-604. doi: 10.1016/j.jtcvs.2017.03.049. Epub 2017 Mar 23 [PubMed PMID: 28528718]
Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circulation research. 2013 Feb 15:112(4):707-20. doi: 10.1161/CIRCRESAHA.112.300853. Epub [PubMed PMID: 23410880]
Dorn C, Perrot A, Grunert M, Rickert-Sperling S. Human Genetics of Tetralogy of Fallot and Double-Outlet Right Ventricle. Advances in experimental medicine and biology. 2024:1441():629-644. doi: 10.1007/978-3-031-44087-8_36. Epub [PubMed PMID: 38884738]
Level 3 (low-level) evidenceMaeda J, Yamagishi H, Furutani Y, Kamisago M, Waragai T, Oana S, Kajino H, Matsuura H, Mori K, Matsuoka R, Nakanishi T. The impact of cardiac surgery in patients with trisomy 18 and trisomy 13 in Japan. American journal of medical genetics. Part A. 2011 Nov:155A(11):2641-6. doi: 10.1002/ajmg.a.34285. Epub 2011 Oct 11 [PubMed PMID: 21990245]
Bell-Cheddar Y, Devine WA, Diaz-Castrillon CE, Seese L, Castro-Medina M, Morales R, Follansbee CW, Alsaied T, Lin JI. Double outlet right ventricle. Frontiers in pediatrics. 2023:11():1244558. doi: 10.3389/fped.2023.1244558. Epub 2023 Sep 25 [PubMed PMID: 37818164]
Samánek M. Boy:girl ratio in children born with different forms of cardiac malformation: a population-based study. Pediatric cardiology. 1994 Mar-Apr:15(2):53-7 [PubMed PMID: 7997413]
Liu A, Diller GP, Moons P, Daniels CJ, Jenkins KJ, Marelli A. Changing epidemiology of congenital heart disease: effect on outcomes and quality of care in adults. Nature reviews. Cardiology. 2023 Feb:20(2):126-137. doi: 10.1038/s41569-022-00749-y. Epub 2022 Aug 31 [PubMed PMID: 36045220]
Level 2 (mid-level) evidenceFarhat N, Vazquez-Jimenez J, Heying R, Seghaye MC. Myocardial mRNA expression of interleukin-6 and hypoxia inducible factor-1α in neonates with congenital cardiac defects. Molecular and cellular pediatrics. 2024 Dec 21:11(1):14. doi: 10.1186/s40348-024-00187-5. Epub 2024 Dec 21 [PubMed PMID: 39708201]
Gordon B, González-Fernández V, Dos-Subirà L. Myocardial fibrosis in congenital heart disease. Frontiers in pediatrics. 2022:10():965204. doi: 10.3389/fped.2022.965204. Epub 2022 Nov 18 [PubMed PMID: 36467466]
Anderson RH, Pickering D, Brown R. Double outlet right ventricle with l-malposition and uncommitted ventricular septal defect. European journal of cardiology. 1975 Aug:3(2):133-42 [PubMed PMID: 1183465]
Sakata R, Lecompte Y, Batisse A, Borromée L, Durandy Y. Anatomic repair of anomalies of ventriculoarterial connection associated with ventricular septal defect. I. Criteria of surgical decision. The Journal of thoracic and cardiovascular surgery. 1988 Jan:95(1):90-5 [PubMed PMID: 3336235]
Lecompte Y, Batisse A, Di Carlo D. Double-outlet right ventricle: a surgical synthesis. Advances in cardiac surgery. 1993:4():109-36 [PubMed PMID: 8252251]
Level 3 (low-level) evidenceLev M, Bharati S, Meng CC, Liberthson RR, Paul MH, Idriss F. A concept of double-outlet right ventricle. The Journal of thoracic and cardiovascular surgery. 1972 Aug:64(2):271-81 [PubMed PMID: 5048382]
Gottschalk I, Abel JS, Menzel T, Herberg U, Breuer J, Gembruch U, Geipel A, Brockmeier K, Berg C, Strizek B. Prenatal diagnosis, associated findings and postnatal outcome of fetuses with double outlet right ventricle (DORV) in a single center. Journal of perinatal medicine. 2019 Apr 24:47(3):354-364. doi: 10.1515/jpm-2018-0316. Epub [PubMed PMID: 30676006]
Lee M, Alahmadi MH, Shahjehan RD. Fontan Completion. StatPearls. 2026 Jan:(): [PubMed PMID: 32644376]
Goo HW. Double Outlet Right Ventricle: In-Depth Anatomic Review Using Three-Dimensional Cardiac CT Data. Korean journal of radiology. 2021 Nov:22(11):1894-1908. doi: 10.3348/kjr.2021.0248. Epub 2021 Sep 13 [PubMed PMID: 34564964]
Macartney FJ, Rigby ML, Anderson RH, Stark J, Silverman NH. Double outlet right ventricle. Cross sectional echocardiographic findings, their anatomical explanation, and surgical relevance. British heart journal. 1984 Aug:52(2):164-77 [PubMed PMID: 6743434]
Hoogerbeets SF, Roest AAW, Valverde I, Gomez-Ciriza G, Kroft L, Hazekamp MG. Printed Models for Better Prediction of Surgery in Patients with Double Outlet Right Ventricle. Pediatric cardiology. 2026 Jan:47(1):201-213. doi: 10.1007/s00246-024-03747-8. Epub 2025 Jan 6 [PubMed PMID: 39762516]
Peek JJ, Bakhuis W, Sadeghi AH, Veen KM, Roest AAW, Bruining N, van Walsum T, Hazekamp MG, Bogers AJJC. Optimized preoperative planning of double outlet right ventricle patients by 3D printing and virtual reality: a pilot study. Interdisciplinary cardiovascular and thoracic surgery. 2023 Aug 3:37(2):. doi: 10.1093/icvts/ivad072. Epub [PubMed PMID: 37202357]
Level 3 (low-level) evidenceWenzel JP, Albrecht JN, Toprak B, Petersen E, Nikorowitsch J, Cavus E, Jahnke C, Riedl KA, Adam G, Twerenbold R, Blankenberg S, Kirchhof P, Lund G, Tahir E, Müllerleile K, Radunski UK. Head-to-head comparison of cardiac magnetic resonance imaging and transthoracic echocardiography in the general population (MATCH). Clinical research in cardiology : official journal of the German Cardiac Society. 2025 May 12:():. doi: 10.1007/s00392-025-02660-1. Epub 2025 May 12 [PubMed PMID: 40353872]
Malakan Rad E, Hijazi ZM. Transcatheter Pulmonary Flow Restrictors: Current Trends and Future Perspectives. Catheterization and cardiovascular interventions : official journal of the Society for Cardiac Angiography & Interventions. 2025 Jan:105(1):165-180. doi: 10.1002/ccd.31308. Epub 2024 Dec 1 [PubMed PMID: 39618067]
Level 3 (low-level) evidencePang K, Yang K, Wang R, Ma K, Xu N, Xing J, Zhang L, Zhang T, Li S. Long-Term Surgical Outcomes in Double Outlet Right Ventricle Based on Detailed Anatomical Sub-Typology. European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery. 2025 Oct 2:67(10):. doi: 10.1093/ejcts/ezaf334. Epub [PubMed PMID: 41056413]
Holten-Andersen M, Lippert M, Holmstrøm H, Brun H, Døhlen G. Current outcomes of live-born children with double outlet right ventricle in Norway. European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery. 2022 Dec 2:63(1):. doi: 10.1093/ejcts/ezac560. Epub [PubMed PMID: 36472441]
Judson JP, Danielson GK, Puga FJ, Mair DD, McGoon DC. Double-outlet right ventricle. Surgical results, 1970-1980. The Journal of thoracic and cardiovascular surgery. 1983 Jan:85(1):32-40 [PubMed PMID: 6848885]
Shen WK, Holmes DR Jr, Porter CJ, McGoon DC, Ilstrup DM. Sudden death after repair of double-outlet right ventricle. Circulation. 1990 Jan:81(1):128-36 [PubMed PMID: 2297820]
Spaeth JP. Perioperative Management of DORV. Seminars in cardiothoracic and vascular anesthesia. 2014 Sep:18(3):281-9. doi: 10.1177/1089253214528048. Epub 2014 Mar 21 [PubMed PMID: 24659409]
Oladunjoye O, Piekarski B, Baird C, Banka P, Marx G, Del Nido PJ, Emani SM. Repair of double outlet right ventricle: Midterm outcomes. The Journal of thoracic and cardiovascular surgery. 2020 Jan:159(1):254-264. doi: 10.1016/j.jtcvs.2019.06.120. Epub 2019 Aug 30 [PubMed PMID: 31597616]