Introduction
Laboratory investigation of hematological disorders typically begins with a complete blood count (CBC) performed using automated hematology analyzers, which provide quantitative data on blood cell number, size, and morphology. Normal red blood cells (RBCs) or erythrocytes are round, biconcave cells with a thin membrane and hemoglobin-rich cytoplasm specialized for oxygen transport. Disorders affecting erythrocytes may reduce production, alter shape or size, or cause qualitative and quantitative hemoglobin abnormalities. Erythrocyte inclusions frequently occur in such conditions and, when correlated with clinical presentation, serve as pathologic evidence of specific diseases.[1] Accurate characterization and identification of these inclusions can establish or support a diagnosis and guide subsequent evaluation.
Issues of Concern
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Issues of Concern
The widespread use of accurate automated hematology analyzers in clinical laboratories has decreased reliance on peripheral blood smear (PBS) evaluation as a primary diagnostic tool. Nevertheless, RBC inclusions are most effectively identified using this modality, offering concise and clinically meaningful clues to the underlying disorder and potentially prompting further investigation. Structural features, composition, and pathophysiological associations of these inclusions provide insight into disease mechanisms.[2] In some cases, significant numbers of erythrocyte inclusions can interfere with automated analyzer data.[3] For example, Heinz bodies and hemoglobin H (HbH) inclusions may yield spurious automated reticulocyte counts.[4]
Causes
Various disorders may induce the development of erythrocyte inclusions, including the following:
- Postsplenectomy asplenia and hyposplenism [5][6]
- Hemoglobinopathies, including thalassemias, unstable hemoglobinopathies, and sickle cell disease (SCD) [7]
- Heavy metal poisoning due to lead, mercury, or arsenic [8]
- Enzymopathies such as glucose-6-phosphate dehydrogenase (G6PD) deficiency [9]
- Anemias, including megaloblastic and sideroblastic forms [10]
- Autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and celiac disease [11][12]
- Neoplastic hematologic disorders, including myelodysplastic syndrome (MDS) and myelofibrosis [13]
- Systemic diseases such as amyloidosis, sarcoidosis, and cirrhosis [14]
Various infectious agents complete part or all of their life cycle within RBCs. These pathogens are not considered true inclusions but appear as intracellular structures on smear. Examples include Plasmodium (malaria) and Babesia (babesiosis) species (see Images. Malaria Parasite Ring Stage in Erythrocytes; Babesiosis). Late normoblasts containing a pyknotic nucleus and reticulocytes containing RNA remnants are considered normal findings.
Clinical Pathology
Most erythrocyte inclusions may be identified and characterized on a routine PBS using standard Romanowsky-type hematological stains, such as Wright or Giemsa. Based on morphology, structural composition, and pathophysiologic correlation, the most common erythrocyte inclusions are categorized as Howell-Jolly bodies, Heinz bodies, Pappenheimer bodies, basophilic stippling, Cabot rings, and inclusions formed by precipitated hemoglobins, such as HbH.[15][16][17]
Morphology
Howell-Jolly Bodies
Howell-Jolly bodies represent residual nuclear DNA within a mature erythrocyte. These inclusions are small, dark, basophilic, smooth, round, punctate cytoplasmic bodies. Howell-Jolly bodies stain purple with Romanowsky dyes, including May-Grünwald-Giemsa, Wright-Giemsa, Leishman, and Diff-Quik® stains (see Image. Howell-Jolly Bodies).
Heinz Bodies
Heinz bodies represent precipitated, denatured hemoglobin deposits within RBCs. These inclusions are seen attached or close to the cellular membrane, may be single or multiple, and are small, round, refractile inclusions measuring 1 to 3 µm in size. Heinz bodies may protrude through the cellular membrane. In a wet blood film, these structures may be observed moving within the cell. Heinz bodies are typically poorly visualized with Romanowsky dyes. When detected, these abnormalities may appear as pink-brown nodular inclusions within RBCs. Heinz bodies stain dark purple-blue and are more readily detected with supravital stains such as methylene blue, brilliant cresyl blue, and methyl violet.[18]
Pappenheimer Bodies
Pappenheimer bodies, also called "siderotic granules," are hemosiderin-containing, small, round, dark, basophilic, or black inclusions situated at the periphery of RBCs. Pappenheimer bodies are not associated with iron-overload conditions but instead result from sideroblastic erythropoiesis, which produces siderotic, nonheme iron intracytoplasmic granules. These granules contain ferric iron, lipids, proteins, and carbohydrates.
Romanowsky dyes help visualize Pappenheimer bodies, which are much smaller than Howell-Jolly bodies. While multiple intracellular inclusions may be present, cells usually contain only a few. When numerous Pappenheimer bodies are present, they can be mistaken for punctate basophilia. These inclusions contain hemosiderin and stain blue with Perls Prussian blue (see Image. Pappenheimer Bodies on Dual Stains). In contrast, punctate basophilia appears pink.[19]
Basophilic Stippling
Basophilic stippling, also called "punctate basophilia," indicates disturbed erythropoiesis, such as defective or accelerated heme synthesis. Basophilic stippling is characterized by numerous irregular basophilic granules dispersed throughout the cytoplasm. The granules may be fine or coarse. Punctate basophilia frequently occurs in disorders involving impaired hemoglobin synthesis and may also result from the production of aberrant, unstable RNA during erythroid maturation. Examples of these disorders include megaloblastic anemia, lead poisoning, and selected hemoglobinopathies. Basophilic stippling is readily demonstrated on PBS analysis using Romanowsky stains (see Image. Basophilic Stippling).
Cabot Rings
Cabot rings are ring-shaped or figure-of-8 inclusions composed of microtubule remnants from the mitotic spindle, nuclear remnants, or abnormal histones.[20] Cabot rings occasionally appear as concentric lines or inclusions with double or multiple loops. These structures stain red, red-purple, or blue-purple with Romanowsky dyes (see Image. Cabot Ring). Cabot rings are most commonly encountered in patients with dyserythropoiesis, such as megaloblastic anemia.
Precipitated Hemoglobin Inclusions
HbH inclusions are precipitated β-chain tetramers observed in α-thalassemia. α-chain inclusions represent α-chain precipitation in β-thalassemia major and appear as irregular nodules near the nucleus of marrow erythroblasts when stained with supravital methyl violet or brilliant cresyl blue.[21]
Mechanisms
RBC inclusions may require differentiation from other intracellular structures due to overlapping morphology. Pappenheimer bodies can resemble the granules seen in basophilic stippling. In contrast to the numerous, diffusely distributed granules of basophilic stippling, Pappenheimer bodies are usually single or few and may be readily distinguished by Perls Prussian blue staining.
Reticulocytes may contain dot-like structures that mimic Pappenheimer bodies. Pappenheimer bodies appear much darker than the reticulofilamentous dots seen in reticulocytes, and Perls staining may be required to confirm the distinction.
HbH may denature and form greenish, round inclusion bodies during reticulocyte staining with new methylene blue. Heinz bodies may also stain pale blue with new methylene blue. Examination of the stained smear under phase-contrast microscopy facilitates differentiation between late reticulocytes and erythrocytes containing inclusion bodies.
Clinicopathologic Correlations
Howell-Jolly Bodies
Howell-Jolly bodies are frequently observed in patients with splenic atrophy or a history of splenectomy. Vitamin B12 deficiency may also result in a small proportion of RBCs containing Howell-Jolly bodies, particularly in chronic or severe deficiency associated with megaloblastosis.
Heinz Bodies
Heinz bodies occur in disorders involving unstable hemoglobin, chemical poisoning, drug toxicity, or G6PD deficiency. These inclusions result from hemoglobin precipitation or denaturation within the RBC. Heinz bodies may also be present in severely unstable hemoglobinopathies that cause hemolytic anemia, with hemoglobin Köln being the most common example.[22] Hemoglobin electrophoresis supports the diagnosis of unstable hemoglobinopathies.
Heinz bodies are also observed in patients who have undergone splenectomy or present with hemolytic anemias caused by acute oxidant-induced hemoglobin denaturation, including G6PD deficiency. When Heinz bodies are detected on PBS assessment, additional testing may include hemoglobin electrophoresis, glutathione stability testing, drug-dependent antibody titers, and methemoglobin and sulfhemoglobin levels.
Basophilic Stippling
Basophilic stippling is observed in various conditions, including lead and other heavy metal poisonings, infections, liver disease, thalassemia, and selected cases of megaloblastic anemia. The presence of basophilic stippling in microcytic, normochromic anemia is highly suggestive of a thalassemia trait.
Both coarse and fine basophilic stippling can occur in patients with lead poisoning. The pathogenesis of this cytologic abnormality in lead toxicity involves the interaction of lead with thiol, carboxylic, and phosphate groups, forming stable complexes with intracellular enzymes. This interaction inhibits pyrimidine 5′-nucleotidase, preventing the removal of clumped or denatured intracellular RNA. The persistence of these RNA aggregates produces the characteristic basophilic stippling seen on peripheral smear.[23] Marked basophilic stippling involving more than 5% of erythrocytes may also occur in congenital pyrimidine 5′-nucleotidase deficiency, an autosomal recessive disorder that causes hereditary nonspherocytic hemolytic anemia.[24]
Pappenheimer Bodies
The presence of Pappenheimer bodies in microcytic anemia suggests sideroblastic erythropoiesis, which may occur in sideroblastic anemia, lead poisoning, or hemochromatosis. Pappenheimer bodies may also form following surgical asplenia. Functional asplenia or hyposplenism in SCD may result in an increased number of erythrocytes containing Pappenheimer bodies. These inclusions are also observed in MDS.
Cabot Rings
Cabot rings are most frequently seen in megaloblastic anemia, MDS, and lead poisoning. These inclusions indicate significant bone marrow stress and are considered a morphologic sign of abnormal erythropoiesis.
Hemoglobin Inclusions
Hemoglobin inclusions are abundant within normoblasts of patients with β-thalassemia and other thalassemias. Patients who have undergone splenectomy may also demonstrate increased numbers of these inclusions on PBS evaluation.
Clinical Significance
Erythrocyte inclusions may provide valuable diagnostic information across a wide spectrum of hematologic disorders. Following a comprehensive medical history and detailed physical examination, targeted evaluation of erythrocyte inclusions can aid in establishing a definitive hematologic diagnosis.
Fatigue is a common presenting symptom of many hematologic conditions, occurring either alone or together with additional systemic manifestations. Concern for anemia frequently leads to an initial laboratory evaluation. Although a CBC is typically informative, a PBS may be necessary when erythrocyte morphology is clinically relevant. Findings from the history and physical examination can guide suspicion toward disorders characterized by erythrocyte inclusions.
Howell-Jolly bodies may be identified on a PBS in patients with conditions such as SCD, asplenia, and MDS. In the U.S., most infants are screened for SCD, although screening may occasionally be missed or yield false results (see Image. Hemoglobin C in Sickle Cell Anemia). Newborn screening for SCD is not routinely performed in many countries. Infants presenting with jaundice or unexplained pain episodes should undergo CBC and PBS evaluation. Detection of Howell-Jolly bodies or sickled erythrocytes should prompt hemoglobin electrophoresis to confirm the diagnosis. Early identification of SCD is critical to prevent complications, including stroke, nephropathy, avascular necrosis of bone, and priapism.[25]
Management options include hydroxyurea and prophylactic penicillin. Newer agents such as crizanlizumab, L-glutamine, and voxelotor have expanded treatment strategies. Hematopoietic stem cell transplantation remains the only potentially curative intervention.[26]
Heinz bodies are observed in various hemoglobinopathies, thalassemias, and G6PD deficiency. Although G6PD deficiency is diagnosed worldwide, it is more prevalent along the malaria belts of Asia, the Mediterranean, sub-Saharan Africa, and the Middle East.[27] The most common form of G6PD deficiency is an inherited enzymopathy, and many patients report a family history of episodic hemolytic anemia. Quantitative spectrophotometric analysis is the gold standard for measuring G6PD activity, whereas qualitative assays may be used as screening tools.
Patients with G6PD deficiency are often asymptomatic unless exposed to oxidative stressors, such as fava beans, certain medications, or infections that precipitate intravascular hemolysis. Pharmacologic triggers include nitrofurantoin, dapsone, primaquine, ribavirin, and rifampin. Hemolysis may also be precipitated by infections caused by hepatitis A, B, or E viruses, cytomegalovirus, or certain streptococcal and staphylococcal species. Once the diagnosis is established, prevention focuses on the avoidance of known triggers. Management of acute hemolysis includes supportive care, folic acid supplementation, and RBC transfusion in cases of severe anemia.[28]
Basophilic stippling may be coarse or fine in appearance. Coarse basophilic stippling is observed in several conditions, including sideroblastic anemia, MDS, lead poisoning, and thalassemia. Fine basophilic stippling may be seen in both health and disease, particularly in conditions characterized by polychromatophilia and increased RBC production.
Basophilic stippling is a frequent PBS finding in lead poisoning.[29][30] Toxicity with this heavy metal most commonly affects children, although adults may also be impacted. Adults with lead toxicity typically present with nonspecific symptoms, such as fatigue and chronic abdominal pain. Early childhood screening for lead poisoning is routinely performed. The Centers for Disease Control and Prevention publishes age-based screening guidelines for all children, with additional guidance for those at increased risk of exposure. Children with lead poisoning may be asymptomatic or present with constipation, headaches, or abdominal pain. At high blood lead levels, clinical manifestations may include appetite loss, drowsiness, impaired concentration, anxiety, and seizures.
Measurement of blood lead levels confirms the diagnosis. Treatment includes chelation therapy and the reduction of exposure by replacing lead-containing materials in paint, construction materials, foods, and water pipes.[31]
Pappenheimer bodies are frequently observed in sideroblastic anemia, lead poisoning, thalassemias, and hemochromatosis.[32] Primary hereditary hemochromatosis is a disorder of iron metabolism. Five types of hereditary hemochromatosis have been described, the most common of which is an autosomal recessive condition. Iron deposition occurs in the liver, heart, skin, pancreas, pituitary gland, and joints.[33] Clinical manifestations of hereditary hemochromatosis include fatigue, arthralgias, and erectile dysfunction. Chronic iron overload may lead to cardiomyopathy, heart failure, cirrhosis, and hepatocellular carcinoma.
The presence of Pappenheimer bodies on a PBS may help narrow the diagnostic differential. Further evaluation of suspected hereditary hemochromatosis includes iron studies and liver enzyme testing. Magnetic resonance imaging of the liver or liver elastography can define the degree of hepatic involvement. Genetic testing is confirmatory.[34] Initial treatment consists of serial phlebotomy, with organ-specific complications of iron overload managed as clinically indicated.
Cabot rings are frequently observed in MDS, myelofibrosis, chronic myeloid leukemia, and megaloblastic anemia. Megaloblastic anemia most commonly results from vitamin B12 or folate deficiency. Insufficiency may arise from inadequate dietary intake, as in strict vegetarian or vegan diets, or from reduced absorption. Celiac disease is one of several disorders that impair absorption of vitamin B12 and iron. Certain medications, inborn errors of metabolism, and pernicious anemia are additional causes of megaloblastic anemia.
Early manifestations of vitamin B12 deficiency include fatigue, glossitis, and angular cheilitis, which are nonspecific findings. Progressive deficiency may result in decreased peripheral position and vibratory sensation, impaired coordination, and gait disturbance. Neuropsychiatric manifestations are late findings.[35] The initial diagnostic step for suspected vitamin B12 deficiency involves determining serum concentrations of this micronutrient. If results are nondiagnostic, measurement of methylmalonic acid and homocysteine levels can aid confirmation. Treatment consists of vitamin B12 replacement by oral or parenteral administration, along with management of the underlying cause.[36]
Media
(Click Image to Enlarge)
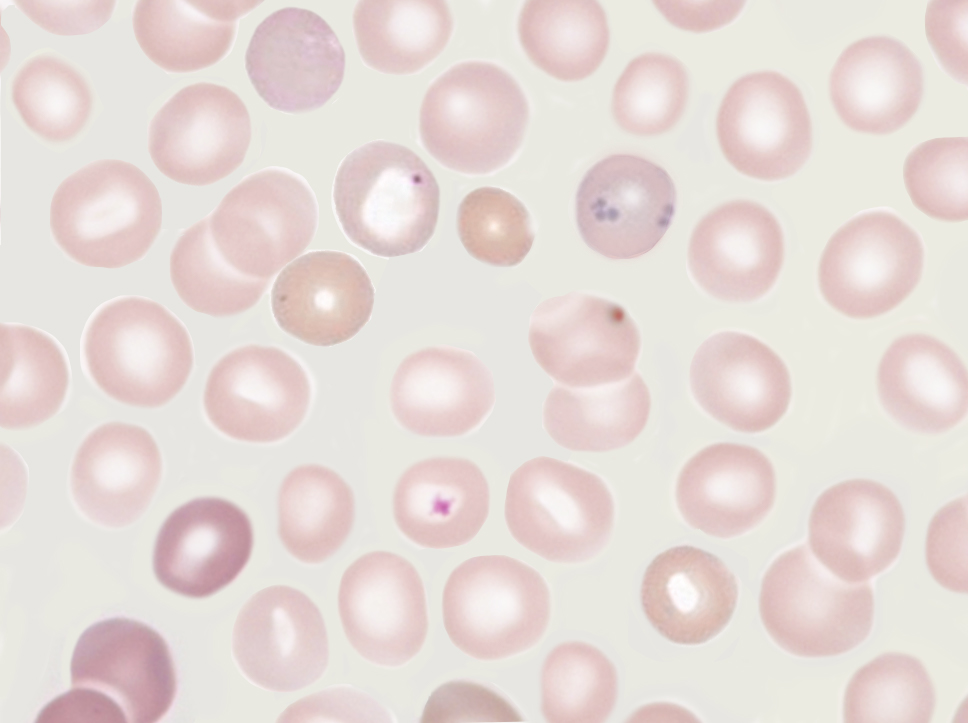
Howell-Jolly Bodies. Howell-Jolly bodies are small, round, basophilic nuclear DNA remnants. These erythrocyte inclusions are observed on peripheral blood smears in patients with absent or impaired splenic function.
Contributed by K Humphreys
(Click Image to Enlarge)
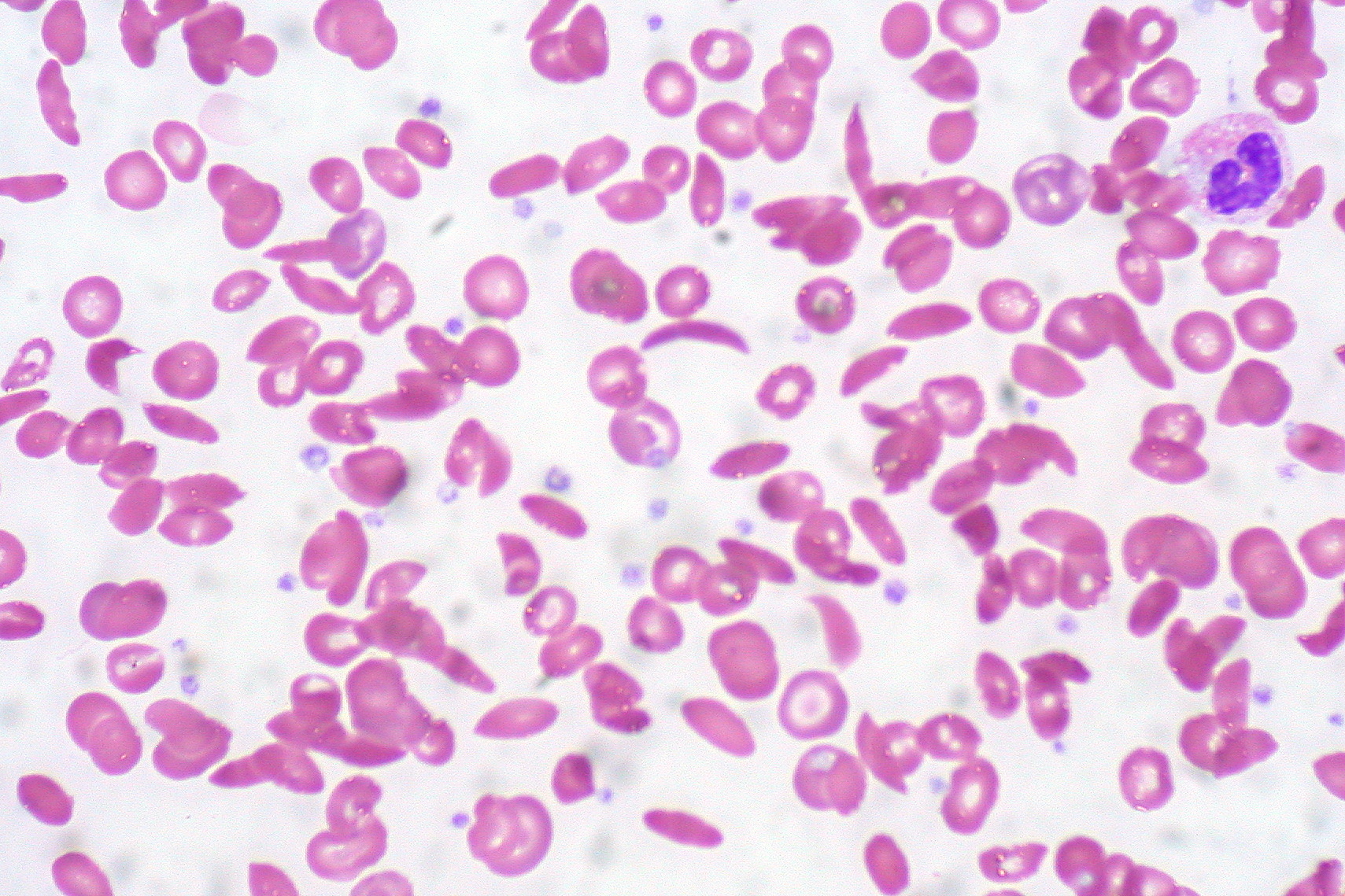
Hemoglobin C in Sickle Cell Anemia. This image shows a peripheral blood smear with numerous abnormally shaped red blood cells characteristic of sickle cell anemia, including sickle-shaped (crescent) cells and target cells associated with hemoglobin C. The altered shapes reflect abnormal hemoglobin that distorts red cell morphology, leading to clinical complications.
Contributed by Ed Uthman (CC BY 2.0 https://creativecommons.org/licenses/by/2.0)
(Click Image to Enlarge)
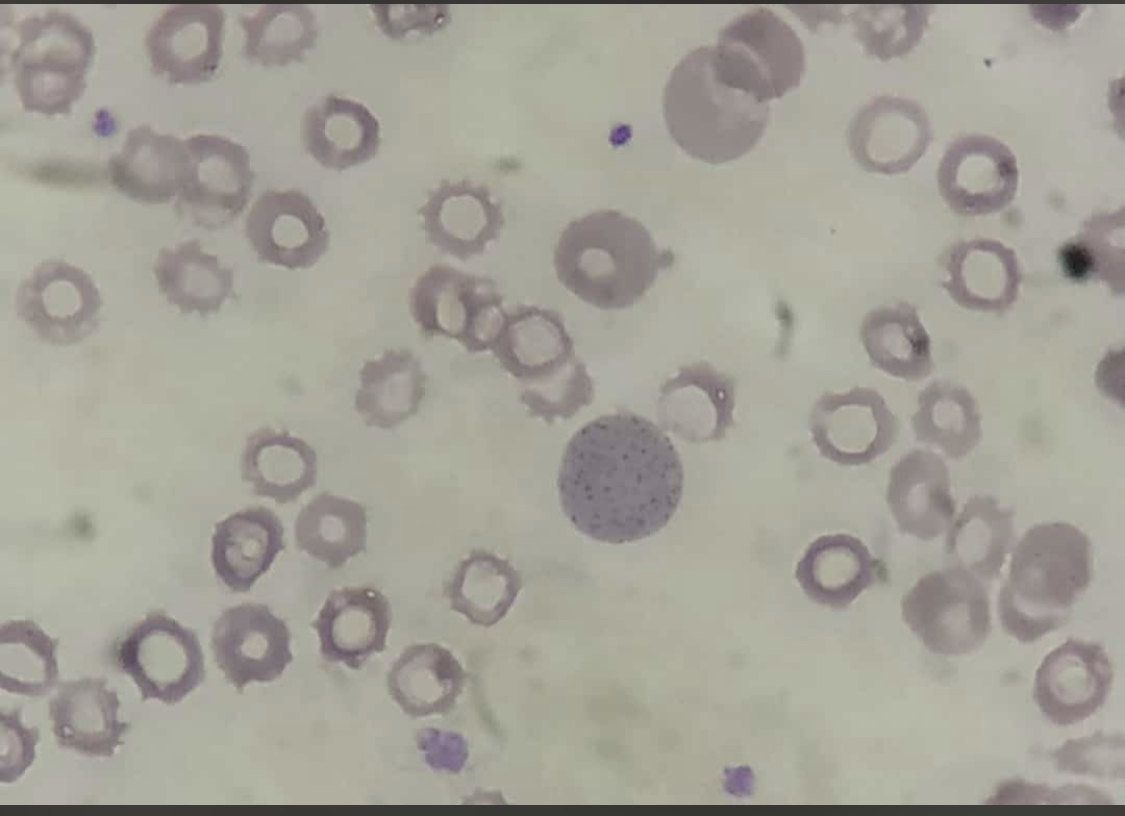
Basophilic Stippling. Red blood cells show coarse, punctate granules dispersed within the cytoplasm, representing abnormal RNA remnants. This finding is commonly associated with lead poisoning, thalassemia, and other disorders of erythropoiesis.
Contributed by AI Khan, MD
(Click Image to Enlarge)
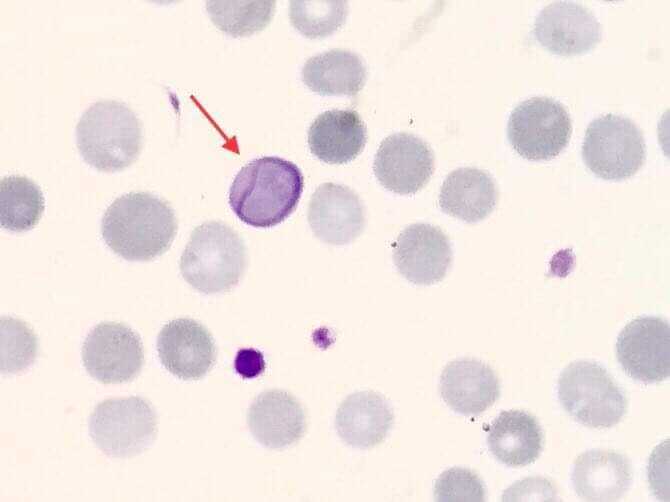
Cabot Ring. A ring-shaped, threadlike inclusion is seen inside an erythrocyte, highlighted by the red arrow, representing remnants of mitotic spindle fibers, often associated with megaloblastic anemia, lead poisoning, or dyserythropoiesis.
"Contributed by Muhammad Zubair, MBBS, FCPS"
(Click Image to Enlarge)
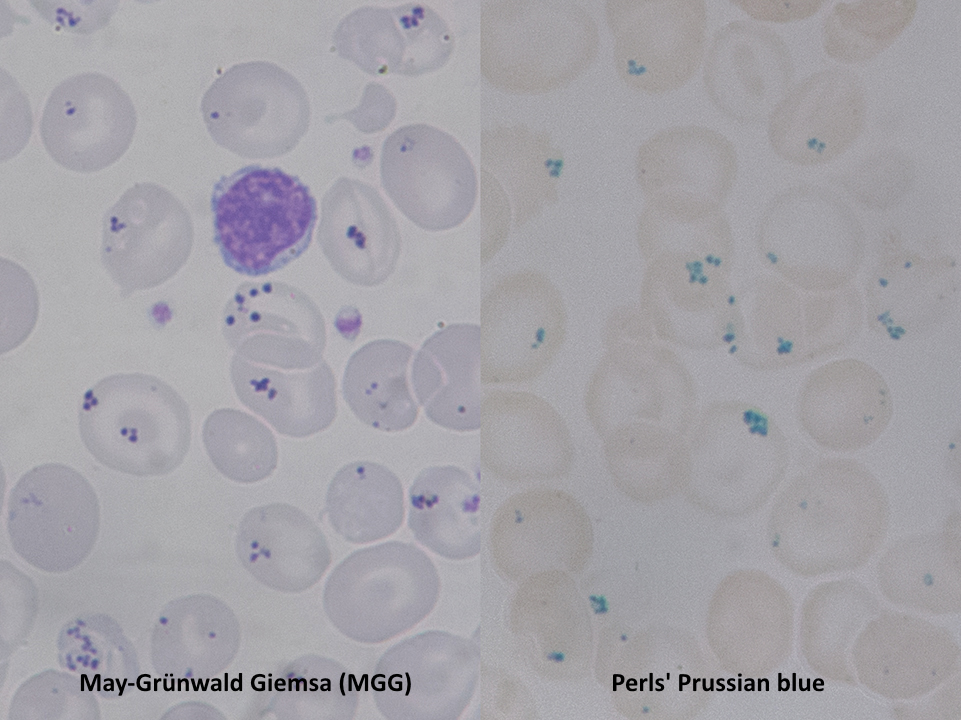
Pappenheimer Bodies on Dual Stains. Red blood cells show small iron-containing inclusions that stain as violet dots with May-Grünwald Giemsa (left) and turn blue with Perls Prussian blue (right), confirming their iron content.
Contributed by Paulo Henrique Orlandi Mourao and licensed under CC BY-SA 4.0. https://creativecommons.org/licenses/by-sa/4.0/?ref=openverse
(Click Image to Enlarge)
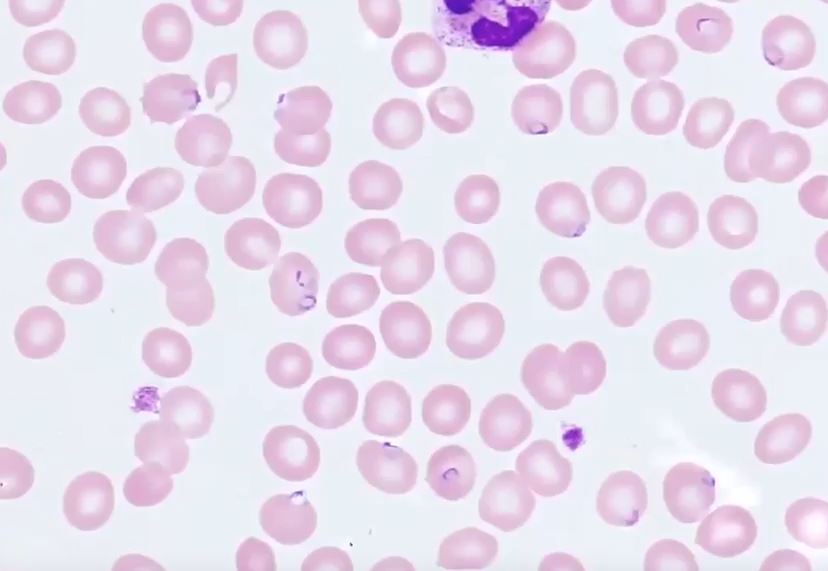
Plasmodium falciparum Ring Stage in Erythrocytes. Multiple ring-shaped trophozoites are visible within red blood cells, a finding characteristic of Plasmodium falciparum infection.
Cade Arries, MD, Public Domain, via Wikimedia Commons
(Click Image to Enlarge)
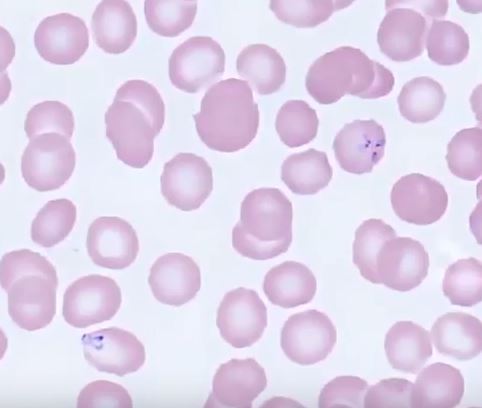
Babesiosis. Multiple intracellular parasites are observed within red blood cells. The upper right corner shows the characteristic Maltese cross formation of 4 parasites.
Image contributed by Dr. Cade Arries, MD, University of Minnesota Medical School
References
Ford J. Red blood cell morphology. International journal of laboratory hematology. 2013 Jun:35(3):351-7. doi: 10.1111/ijlh.12082. Epub 2013 Mar 9 [PubMed PMID: 23480230]
Ford JC, Milner R, Dix DB. Red blood cell morphology reporting: how much is a waste of time? Journal of pediatric hematology/oncology. 2011 Jan:33(1):10-4. doi: 10.1097/MPH.0b013e3181fd6c8b. Epub [PubMed PMID: 21088617]
Lai SK, Yow CM, Benzie IF. Interference of Hb-H disease in automated reticulocyte counting. Clinical and laboratory haematology. 1999 Aug:21(4):261-4 [PubMed PMID: 10583328]
Hinchliffe RF. Errors in automated reticulocyte counts due to Heinz bodies. Journal of clinical pathology. 1993 Sep:46(9):878-9 [PubMed PMID: 8227446]
Level 3 (low-level) evidenceLenti MV, Luu S, Carsetti R, Osier F, Ogwang R, Nnodu OE, Wiedermann U, Spencer J, Locatelli F, Corazza GR, Di Sabatino A. Asplenia and spleen hypofunction. Nature reviews. Disease primers. 2022 Nov 3:8(1):71. doi: 10.1038/s41572-022-00399-x. Epub 2022 Nov 3 [PubMed PMID: 36329079]
Solis M, Perrin J, Guédenet JC, Lesesve JF. [RBCs inclusions after splenectomy: not only Howell-Jolly bodies!]. Annales de biologie clinique. 2013 Mar-Apr:71(2):185-9. doi: 10.1684/abc.2013.0798. Epub [PubMed PMID: 23587584]
Level 3 (low-level) evidenceSpencer-Chapman M, Luqmani A, Layton DM, Bain BJ. Unusual inclusions in hemoglobin H disease post-splenectomy. American journal of hematology. 2018 Jul:93(7):963-964. doi: 10.1002/ajh.25072. Epub 2018 May 6 [PubMed PMID: 29473200]
Sanchez JR, Lynch DT. Histology, Basophilic Stippling. StatPearls. 2026 Jan:(): [PubMed PMID: 31424843]
Herman TF, Killeen RB, Javaid MU. Heinz Body. StatPearls. 2026 Jan:(): [PubMed PMID: 31869086]
Fyfe AJ, Soutar RL. Multiple Pappenheimer bodies. British journal of haematology. 2005 Sep:130(5):651 [PubMed PMID: 16115118]
Level 3 (low-level) evidenceChilds JC, Adelizzi RA, Dabrow MB, Freed N. Splenic hypofunction in systemic lupus erythematosus. The Journal of the American Osteopathic Association. 1994 May:94(5):414-5 [PubMed PMID: 8056631]
Level 3 (low-level) evidenceO'Grady JG, Stevens FM, McCarthy CF. Celiac disease: does hyposplenism predispose to the development of malignant disease? The American journal of gastroenterology. 1985 Jan:80(1):27-9 [PubMed PMID: 3966450]
Tong YT, Nguyen ND, Wahed A. Howell-Jolly Body-Like Inclusions in Neutrophils of Patients With Myelodysplastic Syndrome: A Novel Correlation. Archives of pathology & laboratory medicine. 2019 Jan:143(1):112-114. doi: 10.5858/arpa.2017-0328-OA. Epub 2018 Jul 30 [PubMed PMID: 30059259]
Boyko WJ, Pratt R, Wass H. Functional hyposplenism, a diagnostic clue in amyloidosis. Report of six cases. American journal of clinical pathology. 1982 Jun:77(6):745-8 [PubMed PMID: 7091054]
Level 3 (low-level) evidenceFakoya AO, Amraei R. Histology, Howell Jolly Bodies. StatPearls. 2026 Jan:(): [PubMed PMID: 32491421]
Kano N, Fukui S, Kushiro S, Inui A, Saita M, Kura Y, Sawada U, Naito T. Basophilic stippling in red blood cells in the bone marrow: indication for lead poisoning diagnosis. The Journal of international medical research. 2022 Feb:50(2):3000605221078405. doi: 10.1177/03000605221078405. Epub [PubMed PMID: 35184610]
Erdem N, Berber İ, Aydoğdu İ, Sevinç A. A hundred years after the first article, a recollection: Cabot ring. The Korean journal of internal medicine. 2016 Jan:31(1):199. doi: 10.3904/kjim.2016.31.1.199. Epub 2015 Dec 28 [PubMed PMID: 26767879]
Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Seminars in hematology. 1970 Jul:7(3):341-54 [PubMed PMID: 5425759]
Dubey A, Dey AK, Nandy K, Garg A, Kadakia M. Congenital Sideroblastic Anaemia- Classic Presentation. Journal of clinical and diagnostic research : JCDR. 2016 Sep:10(9):OJ01-OJ02 [PubMed PMID: 27790505]
Bessis M. [Erythrocyte form and deformability for normal blood and some hereditary hemolytic anemias (author's transl)]. Nouvelle revue francaise d'hematologie; blood cells. 1977:18(1):75-94 [PubMed PMID: 896459]
Bain BJ. Alpha chain inclusions in E/beta thalassemia. American journal of hematology. 2008 Nov:83(11):871. doi: 10.1002/ajh.21223. Epub [PubMed PMID: 18553561]
Level 3 (low-level) evidenceMiller DR, Weed RI, Stamatoyannopoulos G, Yoshida A. Hemoglobin Köln disease occurring as a fresh mutation: erythrocyte metabolism and survival. Blood. 1971 Dec:38(6):715-29 [PubMed PMID: 4942314]
de la Serna FJ, Gilsanz F, Ricard P, Urrutia A. [Hemolytic anemia caused by pyrimidine 5'-nucleotidase (P5N) deficiency 15 years later. Apropos of 2 new cases of hereditary deficit and another one of lead poisoning]. Medicina clinica. 1989 Oct 7:93(10):380-2 [PubMed PMID: 2558262]
Level 3 (low-level) evidenceRees DC, Duley JA, Marinaki AM. Pyrimidine 5' nucleotidase deficiency. British journal of haematology. 2003 Feb:120(3):375-83 [PubMed PMID: 12580951]
Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: A Review. JAMA. 2022 Jul 5:328(1):57-68. doi: 10.1001/jama.2022.10233. Epub [PubMed PMID: 35788790]
Brandow AM, Liem RI. Advances in the diagnosis and treatment of sickle cell disease. Journal of hematology & oncology. 2022 Mar 3:15(1):20. doi: 10.1186/s13045-022-01237-z. Epub 2022 Mar 3 [PubMed PMID: 35241123]
Level 3 (low-level) evidenceLuzzatto L, Nannelli C, Notaro R. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematology/oncology clinics of North America. 2016 Apr:30(2):373-93. doi: 10.1016/j.hoc.2015.11.006. Epub [PubMed PMID: 27040960]
Phillips J, Henderson AC. Hemolytic Anemia: Evaluation and Differential Diagnosis. American family physician. 2018 Sep 15:98(6):354-361 [PubMed PMID: 30215915]
Zhao Y, Lv J. Basophilic Stippling and Chronic Lead Poisoning. Turkish journal of haematology : official journal of Turkish Society of Haematology. 2018 Nov 13:35(4):298-299. doi: 10.4274/tjh.2018.0195. Epub 2018 Sep 5 [PubMed PMID: 30182925]
Zamani A, Kazerooni ES, Kasaee SS, Anbardar MH, Mohammadzadeh S, Shekarkhar G, Soleimani N. And the Oscar Goes to Peripheral Blood Film for the Detection of Lead Poisoning in a Complicated Toxic Patient: A Case Report with a Review of Laboratory Clues. Case reports in medicine. 2022:2022():9238544. doi: 10.1155/2022/9238544. Epub 2022 Feb 23 [PubMed PMID: 35251184]
Level 3 (low-level) evidenceMayans L. Lead Poisoning in Children. American family physician. 2019 Jul 1:100(1):24-30 [PubMed PMID: 31259498]
Tyrrell L, Rose G, Shukri A, Kahwash SB. Morphologic changes in red blood cells: An illustrated review of clinically important light microscopic findings. The Malaysian journal of pathology. 2021 Aug:43(2):219-239 [PubMed PMID: 34448787]
Kane SF, Roberts C, Paulus R. Hereditary Hemochromatosis: Rapid Evidence Review. American family physician. 2021 Sep 1:104(3):263-270 [PubMed PMID: 34523883]
Golfeyz S, Lewis S, Weisberg IS. Hemochromatosis: pathophysiology, evaluation, and management of hepatic iron overload with a focus on MRI. Expert review of gastroenterology & hepatology. 2018 Aug:12(8):767-778. doi: 10.1080/17474124.2018.1496016. Epub 2018 Jul 19 [PubMed PMID: 29966105]
Green R, Datta Mitra A. Megaloblastic Anemias: Nutritional and Other Causes. The Medical clinics of North America. 2017 Mar:101(2):297-317. doi: 10.1016/j.mcna.2016.09.013. Epub 2016 Dec 14 [PubMed PMID: 28189172]
Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. American family physician. 2017 Sep 15:96(6):384-389 [PubMed PMID: 28925645]