Introduction
Cogan syndrome is a rare systemic vasculitis characterized by inflammation of the inner ear and eyes.[1] Baumgartner and Morgan first described the condition in 1934 as non–syphilitic interstitial keratitis associated with vestibular dysfunction in a patient with Ménière disease.[2] Dr David G. Cogan later defined it as a distinct clinical entity in 1947.[3] The diagnosis of Cogan syndrome is one of exclusion. This condition typically affects young adults in their second or third decade of life.[4] The proposed pathogenesis involves autoantibodies directed against antigens in the cornea, inner ear, and vascular endothelium.[5]
Cogan syndrome is classified into typical and atypical forms. Typical Cogan syndrome is defined by the onset of ophthalmologic and audiovestibular symptoms within 2 years of each other, usually within 3 to 90 days.[1][6] Atypical Cogan syndrome is diagnosed when this interval exceeds 2 years.[1] Systemic manifestations may include fever, arthralgias, myalgias, arthritis, headache, and aortitis. Cogan syndrome is generally managed with corticosteroids to reduce the frequency and severity of flares and prevent irreversible complications such as sensorineural hearing loss, vestibular dysfunction, blindness, and cardiovascular involvement. Progressive or refractory cases may require biologic or immunomodulatory therapy, and severe cases may need surgical intervention.[1][7][8]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Cogan syndrome was initially thought to have an infectious origin, particularly related to Chlamydia psittaci, based on reports of elevated antibody titers to Chlamydia trachomatis and exposure history in a few patients.[9] However, results from subsequent studies, including those by Vollertsen et al, failed to confirm a causal link.[10] Further investigations have not identified a consistent infectious agent, and antibiotic therapy has been ineffective.[3]
Attention has since shifted to an autoimmune etiology. Patients have demonstrated elevated levels of immunoglobulin G (IgG) and A (IgA) antibodies against human corneal tissue, as well as IgG4 antibodies targeting inner ear tissue.[11] The pathogenic role of these antibodies has been explored using pooled IgG from patients with Cogan syndrome, which reacted with autoantigenic peptides homologous to proteins expressed in the sensory epithelia of the inner ear and in endothelial cells.[5] Passive transfer of these antibodies to mice reproduced features consistent with Cogan syndrome, supporting an autoimmune mechanism.[5]
Infectious or inflammatory triggers may contribute to the initiation of this autoimmune response. Cogan syndrome follows an upper respiratory tract infection episode in 32% to 65% of reported cases.[12] A history of diarrhea, immunization, or dental infection may also precede disease onset.[3][13] Since the onset of the COVID-19 pandemic, 2 cases of atypical Cogan syndrome have been reported following SARS-CoV-2 infection.[12] Additionally, infection may not be the only trigger.
In a recent case series, hemostatic chitosan-tamponade was proposed as a potential trigger after three cases of Cogan syndrome occurred following its use for postpartum hemorrhage.[14] Although causality remains unproven, this association underscores the possible role of immune activation following mucosal or vascular injury. Efforts to identify a specific serological marker for Cogan syndrome have focused on anti-Hsp70 antibodies, which have been detected in some patients with typical Cogan syndrome; however, the finding is inconsistent, particularly in atypical forms.[15] At this time, no single antibody has been established as a definitive diagnostic marker.[3][16]
Epidemiology
Cogan syndrome is a rare disorder, with an estimated prevalence of less than 1 in 500,000 people and approximately 250 cases reported worldwide since its initial description in 1945.[6] The true incidence may be underestimated due to misdiagnosis or incomplete recognition of atypical forms.[3][17][18] In the United States, approximately 60 cases have been documented over the past 62 years.[19] The disease predominantly affects White adults, typically presenting in the second to third decade of life, with a mean age of onset of 25 years (range: 5–63 years).[1][20][21] Although less common in the pediatric population, 55 childhood cases have been reported, with a median age at diagnosis of 12 years.[22] There is no clear sex or racial predominance among reported cases.[1][23]
Pathophysiology
Cogan syndrome is believed to be an autoimmune disorder that targets ocular, inner-ear, and vascular tissues.[5] Histopathologic studies demonstrate lymphocytic and plasma cell infiltration, vascular inflammation, and fibroblast proliferation, consistent with a chronic inflammatory process.[24][25] Further research has investigated the autoimmune hypothesis by analyzing serum samples from patients with Cogan syndrome compared with healthy controls. Elevated levels of IgG and A antibodies against human corneal tissue, as well as IgG antibodies targeting inner ear tissue, have consistently been identified in patients with Cogan syndrome.[11] Results from functional studies have also demonstrated that these IgG antibodies react with autoantigen peptides homologous to proteins expressed in the sensory epithelia of the inner ear and on vascular endothelial cells.[5] In experimental models, passive transfer of patient IgG to mice induced clinical features resembling Cogan syndrome, including cochlear inflammation, vestibular dysfunction, and interstitial keratitis, providing strong evidence that these autoantibodies are pathogenic in disease development.[5]
In the inner ear, antibody-mediated immune responses appear to damage the organ of Corti, spiral ganglion cells, and cochlear neurons, resulting in sensorineural hearing loss, tinnitus, and vestibular symptoms, including vertigo and imbalance.[25][26][27] In ocular tissues, autoimmune mechanisms target corneal stromal cells and endothelium, resulting in mononuclear cell infiltration, corneal neovascularization, and stromal inflammation, which manifest clinically as interstitial keratitis, photophobia, blurred vision, and potential vision loss.[1][8] These immune-mediated processes are more pronounced in typical Cogan syndrome but may also contribute to the ocular involvement seen in atypical Cogan syndrome.
Cogan syndrome is a multiorgan autoimmune disorder that is not limited to ocular or audiovestibular structures.[1] Systemically, autoantibodies may bind to endothelial antigens in medium- and large-sized vessels and cause vascular inflammation, which can result in diffuse vasculitis, aortitis, aneurysm formation, arterial stenosis, and secondary cardiovascular complications.[28][29] Additional systemic symptoms, such as arthralgias, myalgias, and fever, may reflect the presence of circulating immune complexes and endothelial activation in smaller vessels.[22] Together, these studies support a central pathogenic role for autoantibodies in Cogan syndrome, linking molecular immune mechanisms to the clinical triad of inner ear, ocular, and systemic involvement.
Histopathology
Histopathologic examination in Cogan syndrome reveals a chronic inflammatory process affecting multiple tissues, consistent with an autoimmune etiology.[24][25] In the cochlea, there is lymphoplasmacytic infiltration, degeneration of the organ of Corti, loss of spiral ganglion cells, and atrophy of cochlear neurons. Endolymphatic hydrops and secondary bone formation may occur, contributing to progressive sensorineural hearing loss and vestibular dysfunction.[25][26] The vestibulocochlear nerve may exhibit demyelination and atrophy, which correlates clinically with the symptoms of vertigo, imbalance, and tinnitus.[25]
Ocular histopathology predominantly involves the corneal stroma, showing mononuclear cell infiltration, plasma cells, and stromal edema.[1][24] Corneal neovascularization is common, particularly in the posterior stroma near the limbus, and may lead to stromal scarring and vision loss.[1] In atypical Cogan syndrome, inflammation may extend to the iris, ciliary body, retina, or optic nerve, producing uveitis, scleritis, choroiditis, papillitis, or retinal vasculitis.[8][30]
Systemic involvement includes vasculitis of medium- and large-sized vessels.[28][29] Histopathologic examination of affected vessels and valves shows lymphoid cell infiltration, myxomatous changes, fibrinoid necrosis, and intimal thickening.[31][32][33][34][35] In the aorta, these inflammatory changes may result in valvular insufficiency, aneurysm formation, and arterial wall weakening, giving rise to the cardiovascular complications that are the leading cause of mortality in Cogan syndrome.[28] Other organs, including the kidneys and brain, may show evidence of immune-mediated ischemia or infarction, consistent with systemic vasculitis.[36]
History and Physical
The initial presentation of a patient with Cogan syndrome commonly involves either ophthalmologic symptoms (41%) or audiovestibular symptoms (43%), with the latter often mimicking Ménière disease, including vertigo, nausea, vomiting, aural fullness, and tinnitus.[19][27] Ophthalmological symptoms in typical Cogan syndrome present primarily as iritis and non-syphilitic interstitial keratitis (see Image. Interstitial Keratitis, Slit Lamp). In contrast, atypical Cogan syndrome may involve retinal vasculitis, papillitis, central retinal artery occlusion, scleritis, or episcleritis.[8] Around 74% of patients with Cogan syndrome experience eye pain and redness; 50% experience photophobia, tearing, and ocular discomfort; and 42% experience blurred vision.[1]
Cogan syndrome follows an upper respiratory tract infection episode in 32% to 65% of reported cases.[12] Patients may also report a history of diarrhea, immunizations, or dental infections. A recent case series identified hemostatic tamponade using chitosan-based materials as a possible iatrogenic trigger, with three cases of Cogan syndrome developing after patients underwent chitosan tamponade for postpartum hemorrhage.[14]
Physical examination in a patient presenting with Cogan syndrome may reveal spontaneous nystagmus, ataxia, and progressive unilateral or bilateral sensorineural hearing loss (SNHL), which can develop over hours to days and may ultimately progress to total hearing loss. Audiometric evaluation typically demonstrates SNHL across all frequencies, and word recognition ability may vary; electronystagmography often reveals vestibular dysfunction.[37] Ophthalmic examination may reveal ciliary injection, stromal opacities, granular corneal infiltration, and secondary corneal neovascularization, usually affecting both eyes but with possible asymmetry.[30]
As previously stated, Cogan syndrome is subdivided into typical and atypical forms (see Table. Comparison Between Typical and Atypical Cogan Syndrome). Typical Cogan syndrome is defined by the development of ophthalmologic and audiovestibular symptoms within two years of each other, with an average interval of 3 to 90 days. Atypical Cogan syndrome is diagnosed when there is a delay of more than two years between the onset of these symptoms.[1]
Table. Comparison Between Typical and Atypical Cogan Syndrome
| Feature | Typical Cogan Syndrome | Atypical Cogan Syndrome |
| Ocular Involvement | Interstitial keratitis (IK) | Non-IK inflammation (uveitis, scleritis, episcleritis, retinal vasculitis, choroiditis, optic neuritis) |
| Audiovestibular Involvement | Ménière-like vertigo, tinnitus, bilateral sensorineural hearing loss; onset within 2 years of ocular symptoms | Similar audiovestibular symptoms; onset >2 years after ocular symptoms |
| Systemic Involvement | Less common; mild constitutional symptoms (fever, arthralgias, myalgias) | More frequent; severe systemic vasculitis (aortitis, antineutrophil cytoplasmic antibodies-associated vasculitis, glomerulonephritis, neurological involvement) |
| Interval Between Ocular and Audiovestibular Symptoms | Short (<2 years) | Long (>2 years) |
| Prognosis | Better; fewer systemic complications; lower risk of irreversible hearing loss | Worse, higher risk of irreversible hearing loss, vision loss, and cardiovascular complications |
| Treatment Response | Good response to corticosteroids and immunosuppressants | Variable response; often requires aggressive immunosuppression |
Systemic manifestations of Cogan syndrome occur in 30% to 80% of patients and may include fever, weight loss, fatigue, arthralgias, myalgias, and erythromelalgia.[22] Cardiovascular involvement, present in approximately 10% of patients, represents the leading cause of mortality in Cogan syndrome and may manifest as large-vessel vasculitis, aortitis, aortic aneurysm or dissection, aortic valve perforation, and, less commonly, coronary artery involvement leading to myocardial infarction.[28] Neurological symptoms occur in up to 33% of patients; they may include headache, hemiparesis, hemiplegia, aphasia, cerebellar syndrome, epilepsy, meningeal syndrome, or encephalitis secondary to inflammatory effects on medium and large vessels.[1][22]
Gastrointestinal (GI) manifestations, including abdominal pain, hepatitis, and GI hemorrhage, have been reported in up to 22%, 11%, and 16.6% of cases, respectively.[21] Musculoskeletal involvement may include arthritis, myalgias, synovitis, arthralgias, and articular effusion, with muscle biopsies sometimes showing atrophy or necrosis that can mimic myositis.[38] Less common systemic findings include cutaneous lesions (urticarial rash, erythematous nodules, ulcerations, vascular purpura), pulmonary involvement (dyspnea, cough, pleurisy, hemoptysis, thoracic pain, radiographic abnormalities), urinalysis abnormalities, and lymphadenopathy.[1][38]
Additional organs, such as the liver, spleen, kidneys, and urogenital tract, may also be affected. Hepatic involvement may manifest as hepatitis or mild transaminase elevation, while splenic changes can include splenomegaly or reactive lymphoid hyperplasia. Renal involvement may present as proteinuria, hematuria, or glomerulonephritis.[1]
Evaluation
Cogan syndrome remains a diagnosis of exclusion, as there is no definitive biomarker, radiologic finding, or histologic feature that is pathognomonic for the disease.[1] Audiologic evaluation is critical and typically includes pure-tone audiometry, which characteristically reveals SNHL across multiple frequencies, and word recognition testing, which may fluctuate over time. Electronystagmography or videonystagmography can demonstrate vestibular dysfunction, often correlating with clinical vertigo and imbalance.[37] Ophthalmologic evaluation should assess for interstitial keratitis, iritis, uveitis, scleritis, retinal vasculitis, or papillitis, depending on whether the patient has typical or atypical Cogan syndrome. Slit-lamp examination, fundoscopy, and fluorescein angiography can help characterize the extent of ocular involvement.
Laboratory evaluation may reveal nonspecific inflammatory changes, including anemia, leukocytosis, thrombocytosis, elevated erythrocyte sedimentation rate, and elevated C-reactive protein during active disease flares. Hypofibrinogenemia may also be observed in acute episodes. Although antibodies against Chlamydia trachomatis or Chlamydia psittaci may occasionally be detected, such findings are inconsistent and non-diagnostic.[30] Renal involvement can be evaluated with urinalysis, which may reveal proteinuria or hematuria and, in some cases, immune-mediated glomerular injury.
Additional serologic testing may include antinuclear antibodies, antibodies to smooth muscle, lupus anticoagulants, cryoglobulins, anti-neutrophil cytoplasmic antibodies, or rheumatoid factor; these tests are nonspecific and primarily help exclude alternative autoimmune disorders.[30] Antibodies directed against inner-ear or corneal antigens, and against heat shock proteins, may be present in approximately half of patients, but these are not standard diagnostic tests.[15][37]
Imaging is used primarily to evaluate systemic or vascular involvement. Magnetic resonance imaging (MRI) or computed tomography (CT) may identify cerebral ischemic changes or complications from vasculitis. Echocardiography, CT angiography, or MRI angiography can detect aortitis, aneurysms, or valvular abnormalities in patients with cardiovascular symptoms.[28][29] Overall, evaluation relies on a comprehensive history, detailed ophthalmologic and audiovestibular assessment, laboratory testing to exclude alternative diagnoses, and imaging to identify systemic involvement, given the absence of pathognomonic findings for Cogan syndrome.[1]
Treatment / Management
The primary goals of treatment in Cogan syndrome are to reduce the frequency and severity of disease flares; prevent irreversible SNHL, vestibular dysfunction, and ocular complications; and mitigate systemic manifestations, particularly vasculitis.[1] Corticosteroids are the first-line therapy. High-dose corticosteroids, such as intravenous methylprednisolone 500 mg to 1 g daily for 3 days (or oral prednisone ≥1 mg/kg/day), are used to control acute audiovestibular and ocular inflammation. Intratympanic steroid injections may be added to optimize hearing outcomes. Around 37% to 54% of patients with Cogan syndrome may experience persistent hearing loss despite corticosteroid therapy, necessitating escalation to immunosuppressive agents.[1]
Immunosuppressive therapies are employed for refractory or progressive disease. Common agents include methotrexate, azathioprine, cyclophosphamide, cyclosporine A, JAK inhibitors, and rituximab. The choice of agent depends on disease severity, systemic involvement, and patient tolerance, as the rarity of Cogan syndrome limits the availability of randomized controlled trials. Therapy is typically reevaluated after 4 months, with discontinuation or adjustment if no improvement is observed.[1][39][40](B3)
Biologic therapies targeting tumor necrosis factor-α, particularly infliximab, have demonstrated efficacy in patients with resistant disease. A recent review reported clinical remission in 11 of 12 patients treated with infliximab.[41][42] Rituximab has also been shown to reduce systemic inflammation and preserve cochlear function, potentially avoiding cochlear implantation in severe cases.[43](B3)
Cochlear implantation is indicated for patients with severe or treatment-resistant sensorineural hearing loss. Early implantation is preferred to prevent intracochlear fibrosis or ossification, which can compromise outcomes. Post-implantation word recognition scores range from 82.5% to 94%, highlighting the effectiveness of timely surgical intervention.[44] Chronic vestibular dysfunction may be managed with benzodiazepines, antihistamines, or vestibular rehabilitation therapy, tailored to symptom severity.[45] Emerging therapies, including stem-cell-based approaches, are under investigation but require further study to establish long-term safety and efficacy.[46](B2)
Ophthalmologic management involves topical corticosteroids combined with cycloplegics, administered as drops, gels, or ointments (eg, dexamethasone, prednisolone, fluorometholone, hydrocortisone). Treatment duration typically ranges from 6 to 12 months, with close monitoring for steroid-induced glaucoma or cataract formation. Approximately 90% of patients respond favorably, with blindness occurring in 5.5% to 12.7% of cases.[1][40](B3)
Systemic vasculitis, particularly involving large vessels, may require oral cyclophosphamide (2–3 mg/kg/day for 4–6 months) or cyclosporine (≤4 mg/kg/day) if the disease is unresponsive to initial therapy. Surgical intervention for ascending aorta aneurysms or aortic valve disease should generally be deferred until active inflammation is controlled to minimize perioperative risk.[29][40] In all cases, patient education is critical, emphasizing adherence to medical therapy, monitoring for complications, and timely evaluation of auditory, ocular, or systemic changes to optimize outcomes and prevent irreversible organ damage.[1] (B3)
Differential Diagnosis
Diagnosing Cogan syndrome is challenging due to its overlap with other audiovestibular, ophthalmologic, and systemic autoimmune disorders. As a diagnosis of exclusion, careful evaluation to rule out conditions with similar ocular and audiovestibular features is essential.
- Susac syndrome (retinocochleocerebral vasculopathy): SICRET (small infarcts of cochlear, retinal, and encephalic tissues) syndrome is characterized by microvascular occlusions affecting the cochlea, retina, and brain, leading to sensorineural hearing loss, visual disturbances, and neurologic deficits. Unlike Cogan syndrome, lesions are primarily arteriolar, and ocular involvement manifests as branch retinal artery occlusions.[3]
- Congenital syphilis: Interstitial keratitis, sensorineural hearing loss, and Hutchinson teeth (Hutchinson triad) may mimic Cogan syndrome. Serologic testing for syphilis is essential to exclude this diagnosis.[47]
- Vogt-Koyanagi-Harada syndrome: Features of this syndrome include uveitis, sensorineural hearing loss, meningismus, poliosis, and alopecia. Although ocular and auditory findings overlap with Cogan syndrome, the presence of skin depigmentation and alopecia distinguishes this syndrome from Cogan syndrome.[38][48]
- Granulomatosis with polyangiitis: This syndrome is a small-vessel vasculitis that typically involves the ear, eye, respiratory tract, and kidneys. Detection of cytoplasmic anti–neutrophil cytoplasmic antibodies (proteinase 3 antibodies) and characteristic pulmonary and renal involvement can help differentiate granulomatosis with polyangiitis from Cogan syndrome, which does not consistently exhibit these findings.
- Polyarteritis nodosa and Takayasu arteritis: Large- and medium-vessel vasculitides may present with systemic features, including fever, weight loss, myalgias, and aortic involvement. Imaging and serology are critical to distinguish these from Cogan syndrome–associated vasculitides.[17]
- Interstitial keratitis from infectious or autoimmune causes: In addition to syndromic conditions considered in the differential for Cogan syndrome, the differential diagnosis of interstitial keratitis includes infectious etiologies—such as syphilis, chlamydia, leprosy, tuberculosis, brucellosis, leptospirosis, Lyme disease, herpes viruses, measles, mumps, pox viruses, and parasitic infections (eg, onchocerciasis)—as well as autoimmune diseases, including rheumatoid arthritis, sarcoidosis, and granulomatosis with polyangiitis, which may mimic the ocular manifestations of Cogan syndrome.[1][49][1][50][51][52]
Prognosis
The prognosis of Cogan syndrome is highly variable and largely depends on the subtype, the severity of systemic involvement, and the timeliness of intervention. Typical Cogan syndrome generally carries a more favorable prognosis given its relatively limited systemic involvement when compared to atypical Cogan syndrome. In both variants, ophthalmologic symptoms resolve in approximately 90% of patients with treatment, and the prevalence of blindness is around 5.5% to 12.7%.[1]
Auditory outcomes are less favorable than ophthalmologic outcomes. Long-term sensorineural hearing loss may progress to deafness despite corticosteroid or immunosuppressive therapy. In a cohort of 60 patients treated at the Mayo Clinic from 1940 to 2002, complete hearing loss occurred in 1 ear in 18% of patients and in both ears in 52%. Cochlear implantation remains an effective rescue intervention for those with profound hearing loss.[44]
Cardiovascular involvement, though rare, represents the leading cause of mortality in Cogan syndrome. Large-vessel vasculitis may manifest as aortitis, aortic aneurysm or dissection, aortic valve perforation, or coronary artery disease, potentially leading to myocardial infarction or sudden death.[28] Neurologic complications occur in up to 33% of patients, including headache, hemiparesis, hemiplegia, aphasia, cerebellar syndrome, epilepsy, meningeal syndrome, or encephalitis. These manifestations typically result from inflammatory involvement of medium- and large-sized vessels supplying the central nervous system, and the effects may be long-term or permanent.[1][22]
Complications
Cogan syndrome can be complicated by permanent sensorineural hearing loss (50%), blindness (5.5%–12.7%), or complications related to systemic vasculitis.[1] Systemic vasculitis has been reported in 15% to 21% of cases and typically involves large- or medium-sized vessels, although vessels of any size may be affected.[53] Life-threatening cardiovascular complications were reported in 10% of cases, including aortic insufficiency, aneurysms, and dissections.[54]
Other valves may also be involved, leading to complications such as mitral insufficiency.[55] Other organs that may be affected by systemic vasculitis in Cogan syndrome include the kidneys and brain.[36] Cerebrovascular ischemia, including lacunar infarcts and transient ischemic attacks, may result in neurological complications, including aphasia, hemiplegia, and hemiparesis.[56]
Deterrence and Patient Education
Cogan syndrome is a rare autoimmune disorder that presents with audiovestibular and ophthalmologic symptoms, classically nonsyphilitic interstitial keratitis. Because of the disorder's rarity and nonspecific clinical features, which necessitate a diagnosis of exclusion, prompt recognition and treatment are challenging and require strong communication and trust among clinicians, patients, and caregivers. Insufficient treatment or treatment resistance may result in permanent deafness or blindness.
The first line treatment for Cogan syndrome is corticosteroids, though multiple immunosuppressive drugs have been used for more aggressive cases.[29] Cochlear implantation is a surgical option for patients with severe sensorineural hearing loss for whom intensive immunosuppressive regimens are not successful.[29] Patients and caregivers should be educated about the prognosis, management, and expected outcomes of Cogan syndrome to support shared decision-making and optimize clinical outcomes.
Enhancing Healthcare Team Outcomes
Collaboration among the entire healthcare team is mandatory for the management of Cogan syndrome. Primary care clinicians should be educated about this disorder, especially if patients present with progressive hearing loss and eye pain. Referrals to ophthalmology, otolaryngology, neurology, and cardiology are often necessary, particularly in cases of systemic involvement. Screening for other autoimmune diseases, in consultation with rheumatology, should also be considered to improve prognosis and outcomes.
The views expressed in this abstract/manuscript are those of the author(s) and do not reflect the official policy or position of the Department of the Army, Department of Defense, or the United States Government.
Media
(Click Image to Enlarge)
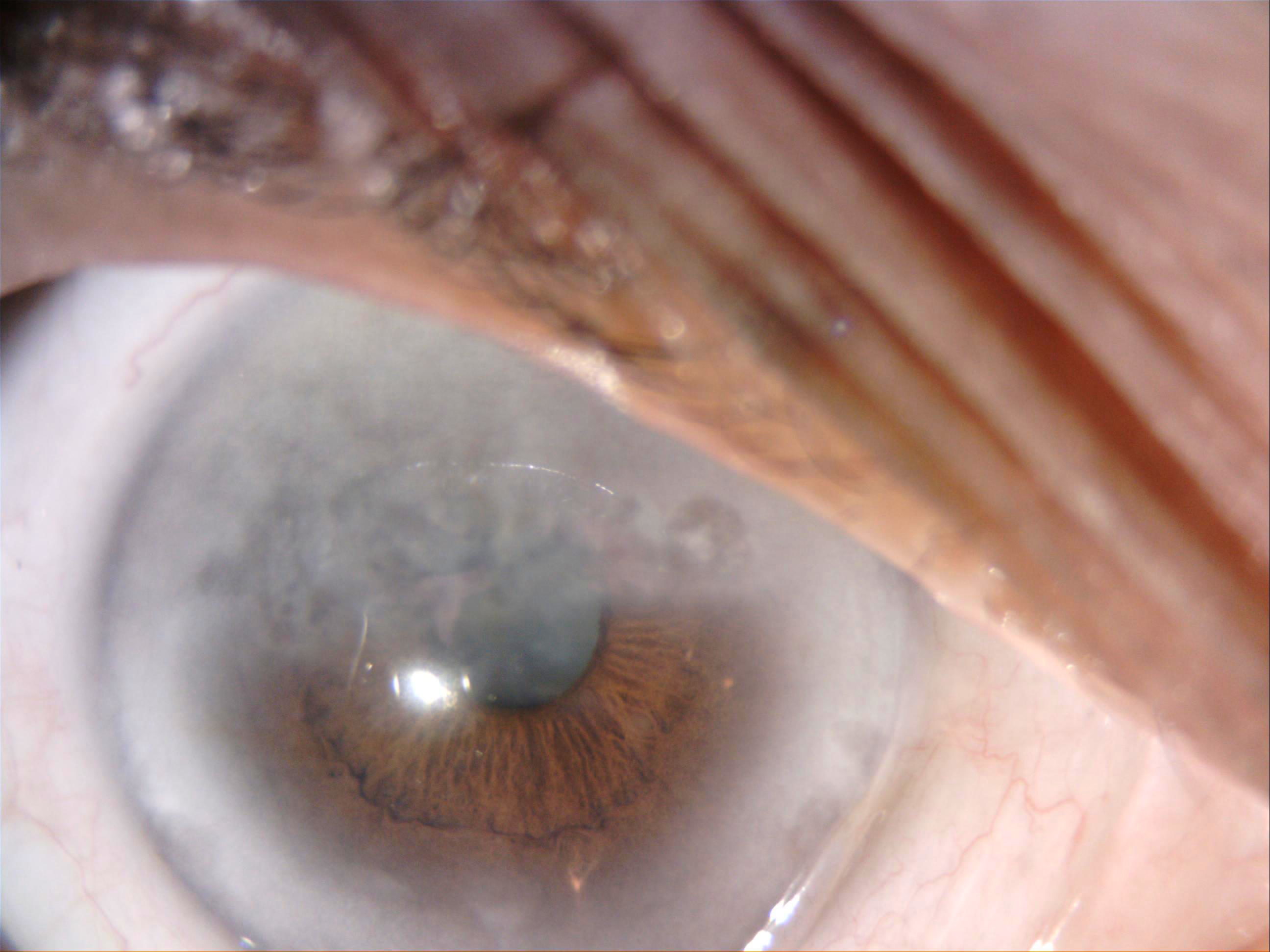
Interstitial Keratitis, Slit Lamp. This slit-lamp image shows interstitial keratitis in a patient with hearing loss; Cogan syndrome was suspected, as the serology for syphilis was negative.
Contributed by P Singh, MD
References
Belfeki N, Ghriss N, Kammoun S, Mekinian A. Cogan's syndrome. A comprehensive review. European journal of internal medicine. 2025 Aug:138():35-43. doi: 10.1016/j.ejim.2025.05.007. Epub 2025 May 17 [PubMed PMID: 40383683]
Cundiff J, Kansal S, Kumar A, Goldstein DA, Tessler HH. Cogan's syndrome: a cause of progressive hearing deafness. American journal of otolaryngology. 2006 Jan-Feb:27(1):68-70 [PubMed PMID: 16360829]
Level 3 (low-level) evidenceIliescu DA, Timaru CM, Batras M, De Simone A, Stefan C. COGAN'S SYNDROME. Romanian journal of ophthalmology. 2015 Jan-Mar:59(1):6-13 [PubMed PMID: 27373108]
Andres T, Lapeyre G, Chan H, Saunier V, Coste V, Touboul D. [Cogan's syndrome]. Journal francais d'ophtalmologie. 2021 Sep:44(7):e419-e421. doi: 10.1016/j.jfo.2020.11.004. Epub 2021 Apr 24 [PubMed PMID: 33902936]
Lunardi C, Bason C, Leandri M, Navone R, Lestani M, Millo E, Benatti U, Cilli M, Beri R, Corrocher R, Puccetti A. Autoantibodies to inner ear and endothelial antigens in Cogan's syndrome. Lancet (London, England). 2002 Sep 21:360(9337):915-21 [PubMed PMID: 12354474]
Level 3 (low-level) evidenceMcCabe BF. Autoimmune sensorineural hearing loss. The Annals of otology, rhinology, and laryngology. 1979 Sep-Oct:88(5 Pt 1):585-9 [PubMed PMID: 496191]
Level 3 (low-level) evidenceEspinoza GM, Wheeler J, Temprano KK, Keller AP. Cogan's Syndrome: Clinical Presentations and Update on Treatment. Current allergy and asthma reports. 2020 Jun 16:20(9):46. doi: 10.1007/s11882-020-00945-1. Epub 2020 Jun 16 [PubMed PMID: 32548646]
Wang Y, Tang S, Shao C, Liu Y. Cogan's syndrome is more than just keratitis: a case-based literature review. BMC ophthalmology. 2023 May 12:23(1):212. doi: 10.1186/s12886-023-02966-6. Epub 2023 May 12 [PubMed PMID: 37173630]
Level 3 (low-level) evidenceHaynes BF, Kaiser-Kupfer MI, Mason P, Fauci AS. Cogan syndrome: studies in thirteen patients, long-term follow-up, and a review of the literature. Medicine. 1980 Nov:59(6):426-41 [PubMed PMID: 6969345]
Vollertsen RS. Vasculitis and Cogan's syndrome. Rheumatic diseases clinics of North America. 1990 May:16(2):433-9 [PubMed PMID: 2189159]
Arnold W, Gebbers JO. [Serum antibodies against corneal and internal ear tissues in Cogan's syndrome]. Laryngologie, Rhinologie, Otologie. 1984 Aug:63(8):428-32 [PubMed PMID: 6384707]
Mezri S, Zitouni C, Sleimi W, Bouzidi M, Sameh S. Case Report: Atypical post-COVID Cogan's syndrome. F1000Research. 2024:13():1104. doi: 10.12688/f1000research.155250.2. Epub 2024 Dec 20 [PubMed PMID: 39931322]
Level 3 (low-level) evidenceMigliori G, Battisti E, Pari M, Vitelli N, Cingolani C. A shifty diagnosis: Cogan's syndrome. A case report and review of the literature. Acta otorhinolaryngologica Italica : organo ufficiale della Societa italiana di otorinolaringologia e chirurgia cervico-facciale. 2009 Apr:29(2):108-13 [PubMed PMID: 20111622]
Level 3 (low-level) evidenceMala N, Zweigart G, Fiedler LS. Multidisciplinary unravelling Cogan's syndrome post-C-section: insights into diagnosis, treatment and a possible identified new trigger. BMJ case reports. 2024 Dec 18:17(12):. pii: e261520. doi: 10.1136/bcr-2024-261520. Epub 2024 Dec 18 [PubMed PMID: 39694649]
Level 3 (low-level) evidenceToubi E. Anti-Hsp70 antibodies and Cogan's syndrome. The Israel Medical Association journal : IMAJ. 2014 May:16(5):311-2 [PubMed PMID: 24979838]
Bonaguri C, Orsoni J, Russo A, Rubino P, Bacciu S, Lippi G, Melegari A, Zavota L, Ghirardini S, Mora P. Cogan's syndrome: anti-Hsp70 antibodies are a serological marker in the typical form. The Israel Medical Association journal : IMAJ. 2014 May:16(5):285-8 [PubMed PMID: 24979832]
Gaubitz M, Lübben B, Seidel M, Schotte H, Gramley F, Domschke W. Cogan's syndrome: organ-specific autoimmune disease or systemic vasculitis? A report of two cases and review of the literature. Clinical and experimental rheumatology. 2001 Jul-Aug:19(4):463-9 [PubMed PMID: 11491507]
Level 3 (low-level) evidenceTirelli G, Tomietto P, Quatela E, Perrino F, Nicastro L, Cattin L, Carretta R. Sudden hearing loss and Crohn disease: when Cogan syndrome must be suspected. American journal of otolaryngology. 2015 Jul-Aug:36(4):590-7. doi: 10.1016/j.amjoto.2015.02.013. Epub 2015 Mar 3 [PubMed PMID: 25841536]
Gluth MB,Baratz KH,Matteson EL,Driscoll CL, Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clinic proceedings. 2006 Apr; [PubMed PMID: 16610568]
Level 2 (mid-level) evidenceTayer-Shifman OE, Ilan O, Tovi H, Tal Y. Cogan's syndrome--clinical guidelines and novel therapeutic approaches. Clinical reviews in allergy & immunology. 2014 Aug:47(1):65-72. doi: 10.1007/s12016-013-8406-7. Epub [PubMed PMID: 24385257]
Level 3 (low-level) evidenceVollertsen RS, McDonald TJ, Younge BR, Banks PM, Stanson AW, Ilstrup DM. Cogan's syndrome: 18 cases and a review of the literature. Mayo Clinic proceedings. 1986 May:61(5):344-61 [PubMed PMID: 3486332]
Level 3 (low-level) evidenceRücklová K, von Kalle T, Koitschev A, Gekeler K, Scheltdorf M, Heinkele A, Blankenburg F, Kötter I, Hospach A. Paediatric Cogan´s syndrome - review of literature, case report and practical approach to diagnosis and management. Pediatric rheumatology online journal. 2023 Jun 8:21(1):54. doi: 10.1186/s12969-023-00830-x. Epub 2023 Jun 8 [PubMed PMID: 37291629]
Level 3 (low-level) evidenceDurtette C, Hachulla E, Resche-Rigon M, Papo T, Zénone T, Lioger B, Deligny C, Lambert M, Landron C, Pouchot J, Kahn JE, Lavigne C, De Wazieres B, Dhote R, Gondran G, Pertuiset E, Quemeneur T, Hamidou M, Sève P, Le Gallou T, Grasland A, Hatron PY, Fain O, Mekinian A, SNFMI and CRI. Cogan syndrome: Characteristics, outcome and treatment in a French nationwide retrospective study and literature review. Autoimmunity reviews. 2017 Dec:16(12):1219-1223. doi: 10.1016/j.autrev.2017.10.005. Epub 2017 Oct 14 [PubMed PMID: 29037902]
Level 2 (mid-level) evidenceFISHER ER, HELLSTROM HR. Cogan's syndrome and systemic vascular disease. Analysis of pathologic features with reference to its relationship to thromboangiitis obliterans (Buerger). Archives of pathology. 1961 Nov:72():572-92 [PubMed PMID: 13893233]
Level 3 (low-level) evidenceRarey KE, Bicknell JM, Davis LE. Intralabyrinthine osteogenesis in Cogan's syndrome. American journal of otolaryngology. 1986 Nov-Dec:7(6):387-90 [PubMed PMID: 3099589]
Level 3 (low-level) evidenceSchuknecht HF, Nadol JB Jr. Temporal bone pathology in a case of Cogan's syndrome. The Laryngoscope. 1994 Sep:104(9):1135-42 [PubMed PMID: 8072362]
Level 3 (low-level) evidenceMarrero-Gonzalez AR, Ward C, Nguyen SA, Jeong SS, Rizk HG. Audiovestibular outcomes in adult patients with cogan syndrome: a systematic review. European archives of oto-rhino-laryngology : official journal of the European Federation of Oto-Rhino-Laryngological Societies (EUFOS) : affiliated with the German Society for Oto-Rhino-Laryngology - Head and Neck Surgery. 2025 Jan:282(1):23-35. doi: 10.1007/s00405-024-08878-5. Epub 2024 Aug 7 [PubMed PMID: 39110231]
Level 1 (high-level) evidenceTakla A, Eid F, Eid MM, Joshi A, Abtahian F, Cheng A, Feitell S. Anterior STEMI in a 25-year-old with Cogan syndrome. Journal of cardiology cases. 2024 Jul:30(1):9-11. doi: 10.1016/j.jccase.2024.02.014. Epub 2024 Mar 5 [PubMed PMID: 39007044]
Level 3 (low-level) evidenceD'Aguanno V, Ralli M, de Vincentiis M, Greco A. Optimal management of Cogan's syndrome: a multidisciplinary approach. Journal of multidisciplinary healthcare. 2018:11():1-11. doi: 10.2147/JMDH.S150940. Epub 2017 Dec 22 [PubMed PMID: 29317827]
Grasland A, Pouchot J, Hachulla E, Blétry O, Papo T, Vinceneux P, Study Group for Cogan's Syndrome. Typical and atypical Cogan's syndrome: 32 cases and review of the literature. Rheumatology (Oxford, England). 2004 Aug:43(8):1007-15 [PubMed PMID: 15150435]
Level 3 (low-level) evidenceCochrane AD, Tatoulis J. Cogan's syndrome with aortitis, aortic regurgitation, and aortic arch vessel stenoses. The Annals of thoracic surgery. 1991 Nov:52(5):1166-7 [PubMed PMID: 1953144]
Level 3 (low-level) evidenceLivingston JZ, Casale AS, Hutchins GM, Shapiro EP. Coronary involvement in Cogan's syndrome. American heart journal. 1992 Feb:123(2):528-30 [PubMed PMID: 1736593]
Level 3 (low-level) evidenceGelfand ML, Kantor T, Gorstein F. Cogan's syndrome with cardiovascular involvement: aortic insufficiency. Bulletin of the New York Academy of Medicine. 1972 May:48(4):647-60 [PubMed PMID: 4503925]
EISENSTEIN B, TAUBENHAUS M. Nonsyphilitic interstitial keratitis and bilateral deafness (Cogan's syndrome) associated with cardiovascular disease. The New England journal of medicine. 1958 May 29:258(22):1074-9 [PubMed PMID: 13552923]
Level 3 (low-level) evidencePinals RS. Cogan's syndrome with arthritis and aortic insufficiency. The Journal of rheumatology. 1978 Fall:5(3):294-8 [PubMed PMID: 748553]
Level 3 (low-level) evidenceKarni A, Sadeh M, Blatt I, Goldhammer Y. Cogan's syndrome complicated by lacunar brain infarcts. Journal of neurology, neurosurgery, and psychiatry. 1991 Feb:54(2):169-71 [PubMed PMID: 2019845]
Level 3 (low-level) evidenceOsborne DC, Jacobson JT, Olsen ML. Cogan's syndrome: auditory and medical management. Journal of the American Academy of Audiology. 1992 May:3(3):225-9 [PubMed PMID: 1581598]
Level 3 (low-level) evidenceNdiaye IC, Rassi SJ, Wiener-Vacher SR. Cochleovestibular impairment in pediatric Cogan's syndrome. Pediatrics. 2002 Feb:109(2):E38 [PubMed PMID: 11826248]
Level 3 (low-level) evidenceMatteson EL, Tirzaman O, Facer GW, Fabry DA, Kasperbauer J, Beatty CW, McDonald TJ. Use of methotrexate for autoimmune hearing loss. The Annals of otology, rhinology, and laryngology. 2000 Aug:109(8 Pt 1):710-4 [PubMed PMID: 10961801]
Allen NB, Cox CC, Cobo M, Kisslo J, Jacobs MR, McCallum RM, Haynes BF. Use of immunosuppressive agents in the treatment of severe ocular and vascular manifestations of Cogan's syndrome. The American journal of medicine. 1990 Mar:88(3):296-301 [PubMed PMID: 2309745]
Level 3 (low-level) evidenceTouma Z, Nawwar R, Hadi U, Hourani M, Arayssi T. The use of TNF-alpha blockers in Cogan's syndrome. Rheumatology international. 2007 Aug:27(10):995-6 [PubMed PMID: 17564714]
Level 3 (low-level) evidenceKalogeropoulos C, Karachalios D, Pentheroudakis G, Tsikou-Papafrangou A, Abou-Asabeh L, Argyropoulou M, Drosos A, Pavlidis N. Development of a low grade lymphoma in the mastoid bone in a patient with atypical Cogan's syndrome: A case report. Journal of advanced research. 2015 May:6(3):523-7. doi: 10.1016/j.jare.2014.05.003. Epub 2014 May 16 [PubMed PMID: 26257951]
Level 3 (low-level) evidenceOrsoni JG, Laganà B, Rubino P, Zavota L, Bacciu S, Mora P. Rituximab ameliorated severe hearing loss in Cogan's syndrome: a case report. Orphanet journal of rare diseases. 2010 Jun 16:5():18. doi: 10.1186/1750-1172-5-18. Epub 2010 Jun 16 [PubMed PMID: 20550723]
Level 3 (low-level) evidenceBoumghit Y, Boucher S, Godey B, Michel G, Bakhos D. Speech reception after cochlear implantation for Cogan's syndrome: Case series following CARE guidelines. European annals of otorhinolaryngology, head and neck diseases. 2023 Sep:140(5):235-238. doi: 10.1016/j.anorl.2023.06.005. Epub 2023 Jul 19 [PubMed PMID: 37479606]
Level 2 (mid-level) evidenceCoelho DH, Lalwani AK. Medical management of Ménière's disease. The Laryngoscope. 2008 Jun:118(6):1099-108. doi: 10.1097/MLG.0b013e31816927f0. Epub [PubMed PMID: 18418279]
Ben-Ami E, Berrih-Aknin S, Miller A. Mesenchymal stem cells as an immunomodulatory therapeutic strategy for autoimmune diseases. Autoimmunity reviews. 2011 May:10(7):410-5. doi: 10.1016/j.autrev.2011.01.005. Epub 2011 Jan 20 [PubMed PMID: 21256250]
Level 3 (low-level) evidenceTripathy K, Sharma YR, Chawla R, Basu K, Vohra R, Venkatesh P. Triads in Ophthalmology: A Comprehensive Review. Seminars in ophthalmology. 2017:32(2):237-250. doi: 10.3109/08820538.2015.1045150. Epub 2015 Jul 6 [PubMed PMID: 26148300]
Stern EM, Nataneli N. Vogt-Koyanagi-Harada Syndrome. StatPearls. 2025 Jan:(): [PubMed PMID: 34662085]
Gyasi ME, Okonkwo ON, Tripathy K. Onchocerciasis. StatPearls. 2025 Jan:(): [PubMed PMID: 32644453]
Singh P, Gupta A, Tripathy K. Keratitis. StatPearls. 2024 Jan:(): [PubMed PMID: 32644440]
Scharl M, Frei P, Fried M, Rogler G, Vavricka SR. Association between Cogan's syndrome and inflammatory bowel disease: a case series. Journal of Crohn's & colitis. 2011 Feb:5(1):64-8. doi: 10.1016/j.crohns.2010.09.003. Epub 2010 Oct 29 [PubMed PMID: 21272808]
Level 3 (low-level) evidenceVavricka SR, Greuter T, Scharl M, Mantzaris G, Shitrit AB, Filip R, Karmiris K, Thoeringer CK, Boldys H, Wewer AV, Yanai H, Flores C, Schmidt C, Kariv R, Rogler G, Rahier JF, ECCO CONFER investigators. Cogan's Syndrome in Patients With Inflammatory Bowel Disease--A Case Series. Journal of Crohn's & colitis. 2015 Oct:9(10):886-90. doi: 10.1093/ecco-jcc/jjv128. Epub 2015 Jul 17 [PubMed PMID: 26188351]
Level 2 (mid-level) evidenceGreco A, Gallo A, Fusconi M, Magliulo G, Turchetta R, Marinelli C, Macri GF, De Virgilio A, de Vincentiis M. Cogan's syndrome: an autoimmune inner ear disease. Autoimmunity reviews. 2013 Jan:12(3):396-400. doi: 10.1016/j.autrev.2012.07.012. Epub 2012 Jul 28 [PubMed PMID: 22846458]
Level 3 (low-level) evidenceSu JW, Low AH, Tay KH, Sebastian MG, Thumboo J, Sin KY. Recurrent aortic aneurysms following thoracic aortic stent-graft repair in a patient with Cogan syndrome. Journal of endovascular therapy : an official journal of the International Society of Endovascular Specialists. 2006 Dec:13(6):779-82 [PubMed PMID: 17154705]
Gasparovic H, Djuric Z, Bosnic D, Petricevic M, Brida M, Dotlic S, Biocina B. Aortic root vasculitis associated with Cogan's syndrome. The Annals of thoracic surgery. 2011 Jul:92(1):340-1. doi: 10.1016/j.athoracsur.2010.12.068. Epub [PubMed PMID: 21718871]
Level 3 (low-level) evidencePanuganti KK, Tadi P, Lui F. Transient Ischemic Attack. StatPearls. 2025 Jan:(): [PubMed PMID: 29083778]